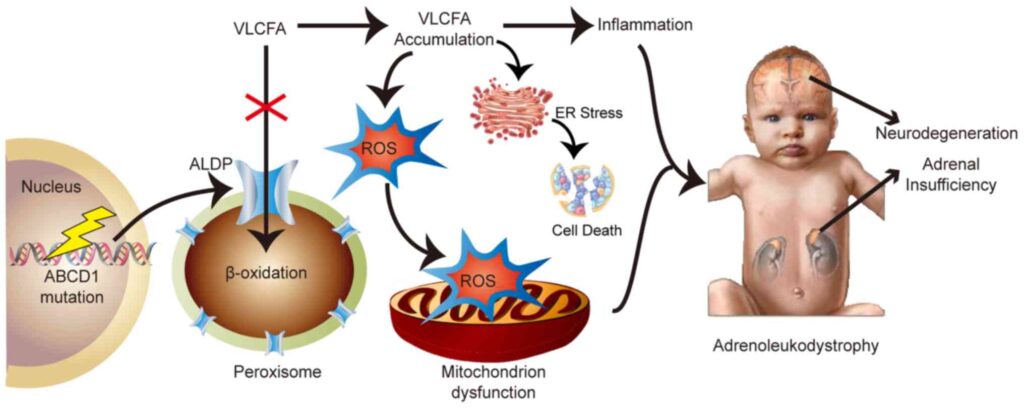
X-linked adrenoleukodystrophy (X-ALD) is a rare, inherited peroxisomal disorder that predominantly affects males. Characterized by the accumulation of very long-chain fatty acids (VLCFAs) in various tissues, it leads to progressive damage of the adrenal cortex, spinal cord, and white matter of the brain. The disorder stems from mutations in the ABCD1 gene, resulting in impaired beta-oxidation of VLCFAs.
Genetic Basis and Inheritance Pattern
X-ALD is inherited in an X-linked recessive manner, meaning the defective ABCD1 gene is located on the X chromosome. Males (XY) with the mutation typically manifest symptoms, whereas females (XX) are usually carriers and may show milder or late-onset signs.
Pathophysiology: VLCFA Accumulation and Cellular Dysfunction
In individuals with X-ALD, a defective ABCD1 gene leads to malfunctioning of peroxisomal ATP-binding cassette (ABC) transporter proteins. This results in the intracellular buildup of VLCFAs—primarily hexacosanoic acid (C26:0)—in:
- Adrenal cortex: leading to adrenal insufficiency (Addison’s disease)
- Central nervous system: resulting in demyelination of cerebral white matter
- Spinal cord: causing adrenomyeloneuropathy (AMN)
The accumulation of VLCFAs disrupts membrane integrity, induces oxidative stress, and triggers inflammatory demyelination, culminating in severe neurological deterioration.
Clinical Variants of X-Linked Adrenoleukodystrophy
X-ALD manifests in several clinical phenotypes, which may vary in onset, severity, and progression.
1. Childhood Cerebral ALD
- Onset: Ages 4–10
- Features: Behavioral changes, progressive cognitive decline, visual and auditory impairment, spasticity, seizures
- Rapid progression, often fatal within a few years without intervention
2. Adrenomyeloneuropathy (AMN)
- Onset: Adulthood (20s–40s)
- Features: Progressive stiffness and weakness in legs, urinary disturbances, sexual dysfunction, peripheral neuropathy
- Gradual progression with variable adrenal involvement
3. Addison-Only Phenotype
- Onset: Childhood or adolescence
- Features: Primary adrenal insufficiency without neurological symptoms
- May evolve into cerebral ALD or AMN over time
4. Symptomatic Female Carriers
- 20–50% of carriers develop AMN-like symptoms later in life
- Often milder with slower progression
Signs and Symptoms by System Involvement
| System | Clinical Manifestations |
|---|---|
| Neurological | Hyperactivity, learning difficulty, vision loss, spasticity |
| Adrenal | Fatigue, hypotension, hyperpigmentation, salt craving |
| Musculoskeletal | Stiffness, gait disturbance, muscle weakness |
| Urogenital | Erectile dysfunction, urinary incontinence |
| Behavioral | Personality changes, attention deficit, emotional lability |
Diagnostic Strategies for X-Linked Adrenoleukodystrophy
1. Biochemical Testing
- Plasma VLCFA levels: Elevated concentrations of C24:0 and C26:0
- Ratio of C24:0/C22:0 and C26:0/C22:0: Diagnostic marker
2. Genetic Testing
- Detection of mutations in the ABCD1 gene through sequencing
- Carrier testing and prenatal diagnosis available
3. MRI Brain Imaging
- Symmetric demyelination in parieto-occipital regions in cerebral ALD
- Gadolinium enhancement indicates active inflammation
4. Adrenal Function Testing
- Low cortisol and elevated ACTH levels confirm adrenal insufficiency
Newborn Screening and Early Detection
Several regions now include X-ALD in newborn screening panels due to its prevalence and therapeutic potential when detected early. Early identification allows pre-symptomatic monitoring and timely intervention.
Treatment and Management Approaches
1. Hematopoietic Stem Cell Transplantation (HSCT)
- Most effective when performed early in cerebral ALD
- Slows or halts demyelination progression
- High-risk procedure requiring careful selection
2. Adrenal Hormone Replacement
- Lifelong glucocorticoid and mineralocorticoid supplementation for adrenal insufficiency
3. Gene Therapy
- Emerging option using lentiviral vectors to correct ABCD1 defects
- Shown promise in halting disease progression in early trials
4. Lorenzo’s Oil
- Mixture of oleic and erucic acid
- Reduces VLCFA levels but does not reverse neurological damage
- Preventive role in asymptomatic boys
5. Symptomatic and Supportive Care
- Physical therapy, antispasticity medications, occupational therapy
- Assistive devices for mobility and daily living
Prognosis and Long-Term Outcomes
- Cerebral ALD: Rapid deterioration if untreated; HSCT may improve survival
- AMN: Slowly progressive; quality of life impacted by gait and bladder issues
- Addison-only: Manageable with hormone therapy but risk of neurological evolution remains
- Prognosis greatly improves with early diagnosis and treatment
Ongoing Research and Clinical Trials
Research continues into:
- Advanced gene therapy techniques (e.g., CRISPR-based correction)
- Anti-inflammatory and neuroprotective agents
- Disease-modifying agents to halt VLCFA accumulation
- Enhanced screening methods for early detection
Frequently Asked Questions:
Q1: Is X-linked adrenoleukodystrophy curable?
There is no complete cure. Early treatment with stem cell transplant or gene therapy can stabilize the disease.
Q2: What causes X-ALD?
Mutations in the ABCD1 gene impair peroxisomal function, leading to VLCFA buildup.
Q3: Can females have X-ALD?
Females are typically carriers but can develop mild symptoms later in life.
Q4: Is Lorenzo’s Oil effective?
It may lower VLCFA levels in asymptomatic patients but is not a cure for symptomatic forms.
Q5: Can newborns be tested for X-ALD?
Yes, newborn screening is available in several regions and helps initiate early intervention.
X-linked adrenoleukodystrophy is a devastating but increasingly manageable disorder. With advancements in diagnostic techniques, early detection through newborn screening, and evolving therapeutic options including gene therapy and HSCT, we now have the potential to alter the trajectory of this disease. Continued research and awareness are paramount in delivering hope and improved quality of life for affected individuals and their families.