Systemic dermatomyositis is a chronic, idiopathic inflammatory myopathy characterized by distinctive cutaneous manifestations and progressive symmetrical proximal muscle weakness. As a multisystem autoimmune disease, it predominantly affects adults and children alike, with particular prevalence among women aged 40–60 years. Early recognition and a multidisciplinary approach to treatment significantly influence long-term outcomes and reduce morbidity.
Pathogenesis and Immunological Mechanisms of Dermatomyositis
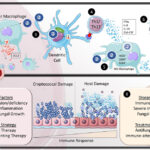
Dermatomyositis is mediated by immune complex deposition and complement activation targeting the microvasculature of skin and muscle. The pathophysiology involves:
- Complement-mediated capillary injury
- B-cell and T-cell dysregulation
- Upregulation of interferon-inducible genes (type I interferon signature)
- Microangiopathy affecting perifascicular muscle fibers
Clinical Manifestations of Systemic Dermatomyositis
Cutaneous Features
Dermatomyositis is distinguished by hallmark skin lesions, which may precede, accompany, or follow muscle symptoms:
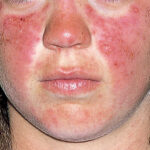
- Heliotrope Rash: Violet or dusky discoloration around the eyelids with periorbital edema
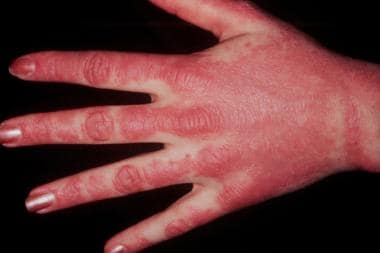
- Gottron’s Papules: Violaceous papules over knuckles, elbows, and knees
- Shawl Sign: Photosensitive rash over the shoulders, upper back, and neck
- Mechanic’s Hands: Hyperkeratotic, fissured palms resembling manual labor
Musculoskeletal Involvement
- Proximal muscle weakness: Difficulty climbing stairs, rising from a chair, or lifting objects
- Myalgia: Often present, but less prominent than weakness
- Dysphagia: Esophageal involvement due to pharyngeal muscle weakness
Systemic Complications
- Interstitial lung disease (ILD)
- Cardiac involvement: Arrhythmias, myocarditis
- Malignancy association: Increased risk in adults, especially within 3–5 years of diagnosis
Diagnostic Criteria and Laboratory Evaluation
Key Diagnostic Tools
| Test/Tool | Findings |
|---|---|
| Serum Creatine Kinase (CK) | Elevated in active muscle disease |
| Electromyography (EMG) | Myopathic changes, fibrillation potentials |
| Muscle Biopsy | Perifascicular atrophy, perivascular inflammation |
| Autoantibody Profile | Anti-Mi-2, Anti-MDA5, Anti-TIF1-γ, Anti-NXP2, Anti-SAE |
| MRI of Skeletal Muscles | Edema and inflammation in proximal muscle groups |
| Skin Biopsy (if indicated) | Interface dermatitis with mucin deposition |
Bohan and Peter Diagnostic Criteria (Modified)
- Symmetrical proximal muscle weakness
- Elevated serum muscle enzymes
- Myopathic EMG changes
- Positive muscle biopsy
- Classic dermatomyositis rash
Diagnosis requires the rash plus at least three additional criteria.
Subtypes and Autoantibody Profiles
Classic Dermatomyositis
- Cutaneous and muscle involvement
- Often responsive to corticosteroids
Clinically Amyopathic Dermatomyositis (CADM)
- Cutaneous manifestations without muscle weakness for 6 months
- Associated with anti-MDA5 antibodies and ILD
Juvenile Dermatomyositis
- Onset before 18 years of age
- Higher risk of calcinosis and GI vasculopathy
Paraneoplastic Dermatomyositis
- Strongly associated with internal malignancies
- Linked with anti-TIF1-γ autoantibodies
Comprehensive Treatment Strategies
Corticosteroids: First-Line Immunosuppression
- Prednisone 1 mg/kg/day, tapered over several months based on response
- IV methylprednisolone pulses in severe or refractory cases
Immunosuppressive Agents
Often added to minimize steroid toxicity and achieve remission:
- Methotrexate: Weekly dosing, effective in both muscle and skin disease
- Azathioprine: Particularly useful in ILD-associated disease
- Mycophenolate mofetil: Effective in steroid-sparing regimens and ILD
- Cyclophosphamide: Reserved for life-threatening systemic involvement
Biologic Therapies and Novel Agents
- Rituximab (anti-CD20): Effective in refractory dermatomyositis
- IVIG: Beneficial in rapidly progressive cases or dysphagia
- Janus kinase (JAK) inhibitors: Investigational use for interferon-driven subtypes
- Anti-MDA5 antibody-specific management: High-dose immunosuppressants and close monitoring of ILD
Malignancy Screening in Dermatomyositis
Owing to the paraneoplastic nature of the disease in some individuals, especially those with anti-TIF1-γ positivity, age-appropriate malignancy workup is mandatory.
Recommended Screening Modalities
- CT chest, abdomen, pelvis
- Mammography or breast MRI
- Pelvic ultrasound
- Colonoscopy
- PSA testing in men
- Whole-body PET-CT (in selected high-risk cases)
Prognosis and Monitoring
Favorable Prognostic Indicators
- Early diagnosis and initiation of immunosuppressive therapy
- Absence of ILD or malignancy
- Positive response to corticosteroids
Poor Prognostic Factors
- Anti-MDA5 positivity
- Severe ILD or cardiac involvement
- Delayed treatment initiation
- Malignancy-associated dermatomyositis
Monitoring Protocol
| Interval | Evaluation |
|---|---|
| Every 4–6 weeks | CK levels, strength assessment |
| Every 3–6 months | Pulmonary function tests if ILD |
| Annually | Cancer screening, full lab workup |
Frequently Asked Questions:
Q1. What triggers systemic dermatomyositis?
While the precise cause is unknown, genetic predisposition and environmental triggers such as infections or malignancy play contributory roles.
Q2. Is dermatomyositis curable?
It is not curable but highly manageable with immunosuppressive and supportive therapies. Early intervention improves outcomes.
Q3. What are the risks of untreated dermatomyositis?
Progressive muscle weakness, permanent disability, ILD, and increased risk of malignancy.
Q4. Can dermatomyositis occur without muscle weakness?
Yes. This form is called clinically amyopathic dermatomyositis (CADM), where skin symptoms predominate.
Q5. How is interstitial lung disease managed in dermatomyositis?
With immunosuppressants like mycophenolate or cyclophosphamide, and close pulmonary monitoring.