Small lymphocytic lymphoma (SLL) with 17p deletion represents a high-risk subset of this indolent B-cell non-Hodgkin lymphoma. The loss of the short arm of chromosome 17, which harbors the TP53 tumor suppressor gene, profoundly alters the disease course, significantly impacting response to conventional therapies and long-term outcomes. Understanding the genetic implications of this deletion is critical to optimizing management strategies for affected patients.
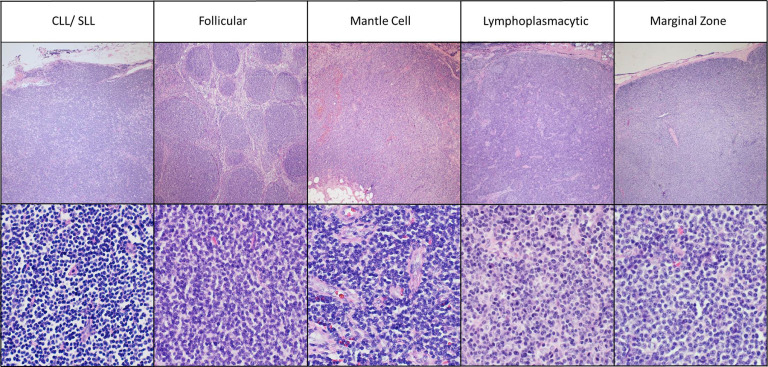
17p Deletion and TP53 Dysfunction in SLL
The 17p deletion in SLL removes a portion of chromosome 17 containing the TP53 gene, a vital regulator of apoptosis and DNA repair. TP53 dysfunction is associated with treatment resistance, rapid progression, and inferior survival rates in both SLL and chronic lymphocytic leukemia (CLL). Given the molecular and clinical overlap between SLL and CLL, findings are largely extrapolated across both conditions.
Biological Consequences
- Impaired apoptosis leading to treatment resistance
- Genomic instability and clonal evolution
- Increased risk of Richter’s transformation
- Reduced effectiveness of DNA-damaging agents such as fludarabine
Clinical Features of SLL with 17p Deletion
Patients with 17p-deleted SLL often present with more aggressive clinical features compared to typical SLL cases. These may include:
- Rapidly enlarging lymphadenopathy
- B symptoms (fever, night sweats, weight loss)
- Shorter time to first treatment
- Resistance to traditional chemoimmunotherapy
- Elevated serum β2-microglobulin and lactate dehydrogenase (LDH)
Diagnostic Approach to Detect 17p Deletion in SLL
Identification of 17p deletion is essential for risk stratification and treatment planning. Testing is recommended at diagnosis and prior to initiating therapy.
Essential Diagnostic Tools
- FISH (Fluorescence In Situ Hybridization): Gold standard for detecting 17p deletion
- Next-Generation Sequencing (NGS): Identifies accompanying TP53 mutations not detectable by FISH
- Flow Cytometry and Immunophenotyping: Confirms SLL diagnosis and helps distinguish from other lymphomas
- Bone Marrow Biopsy: May be used to assess disease extent
Prognostic Implications of 17p Deletion
The presence of 17p deletion or TP53 mutation is associated with:
- Median progression-free survival: Significantly shorter compared to TP53-intact SLL
- Overall survival: Poorer outcomes, often under five years despite treatment
- Richter’s transformation: Higher incidence among this high-risk group
Risk-adapted treatment strategies are crucial to improving prognosis.
First-Line Treatment of SLL with 17p Deletion
Traditional chemoimmunotherapy (e.g., fludarabine, cyclophosphamide, rituximab – FCR) is ineffective in the setting of 17p deletion and is no longer recommended. The therapeutic focus has shifted to targeted therapies that bypass the need for intact TP53 function.
Preferred First-Line Agents
BTK Inhibitors
- Ibrutinib (first-in-class): Proven efficacy as monotherapy in 17p-deleted SLL
- Acalabrutinib: Second-generation BTK inhibitor with favorable tolerability
- Continuous therapy is often required due to risk of relapse on discontinuation
BCL-2 Inhibitor – Venetoclax
- Effective in TP53-deficient SLL
- Usually combined with anti-CD20 antibody (e.g., Obinutuzumab)
- Fixed-duration regimens allow treatment-free remission in many cases
Combination and Sequential Strategies
Optimal sequencing and combination of targeted agents continue to evolve. In high-risk SLL, combining BTK inhibitors with venetoclax or switching upon intolerance is common in clinical practice.
Emerging Combinations:
- Ibrutinib + Venetoclax
- Acalabrutinib + Venetoclax + Obinutuzumab
Such combinations aim to achieve deeper remissions and potentially overcome resistance mechanisms.
Salvage Therapy and Clinical Trials
In relapsed or refractory 17p-deleted SLL, especially post-BTK or BCL-2 failure, clinical trial enrollment is strongly advised. Options include:
- CAR T-cell therapy: Autologous anti-CD19 CAR T-cells showing promise in high-risk B-cell lymphomas
- Non-covalent BTK inhibitors: Agents like pirtobrutinib show efficacy in BTK-resistant disease
- Bispecific antibodies: Experimental therapies targeting CD20 and CD3
Monitoring and Follow-Up in High-Risk SLL
Given the aggressive biology, patients with 17p-deleted SLL require close monitoring even during periods of remission.
Recommended Surveillance:
- Regular CBCs and metabolic panels
- Imaging as clinically indicated (CT or PET)
- Molecular reassessment of 17p deletion and TP53 mutations prior to each new line of therapy
- Vigilance for Richter’s transformation
Frequently Asked Questions:
What is the significance of 17p deletion in SLL?
It indicates high-risk disease with poor response to standard chemotherapy and requires treatment with novel targeted agents.
Can patients with 17p-deleted SLL achieve long-term remission?
Yes, especially with newer agents like BTK and BCL-2 inhibitors. Fixed-duration regimens and combination strategies show promising durability.
Is FCR effective in 17p-deleted SLL?
No. Due to impaired TP53 function, these patients are largely resistant to fludarabine-based chemotherapy.
What are the best treatment options for 17p-deleted SLL?
Preferred first-line therapies include BTK inhibitors (e.g., ibrutinib, acalabrutinib) or venetoclax-based regimens combined with anti-CD20 antibodies.
Is 17p deletion the same as a TP53 mutation?
Not always. Deletion involves loss of the TP53 gene region, while mutation affects gene function. Both confer similar high-risk features.
Small lymphocytic lymphoma with 17p deletion is a distinct and challenging subtype that requires a precision medicine approach. Identification of the 17p deletion at diagnosis and before each treatment decision is critical. Novel targeted therapies, particularly BTK and BCL-2 inhibitors, offer hope in managing this aggressive disease subset, though ongoing research and clinical trials are vital for improving outcomes further.