Processing-deficient progeroid laminopathy linked to a heterozygous LMNA mutation is a rare genetic disorder marked by premature aging features and multisystem involvement. The disorder arises from defects in the post-translational processing of lamin A, a key structural protein of the nuclear envelope encoded by the LMNA gene. The pathological consequence is a toxic buildup of abnormal prelamin A, leading to nuclear instability and tissue degeneration reminiscent of progeria syndromes.
Genetic Basis: The Role of LMNA Mutations
The LMNA gene encodes two major splice variants, lamin A and lamin C, which are integral to nuclear structure, gene expression regulation, and chromatin organization. A heterozygous mutation in LMNA, particularly those affecting the cleavage of prelamin A by ZMPSTE24 (a zinc metalloproteinase), results in the accumulation of farnesylated prelamin A.
This unprocessed or partially processed form is highly cytotoxic, disrupting nuclear integrity and driving premature cellular senescence.
Molecular Mechanism of Disease Progression
Key Events in Laminopathy Pathogenesis
- LMNA Mutation → aberrant prelamin A protein
- Defective ZMPSTE24-mediated processing
- Prelamin A accumulation at the nuclear envelope
- Nuclear deformation, genomic instability
- Cellular senescence and tissue dysfunction
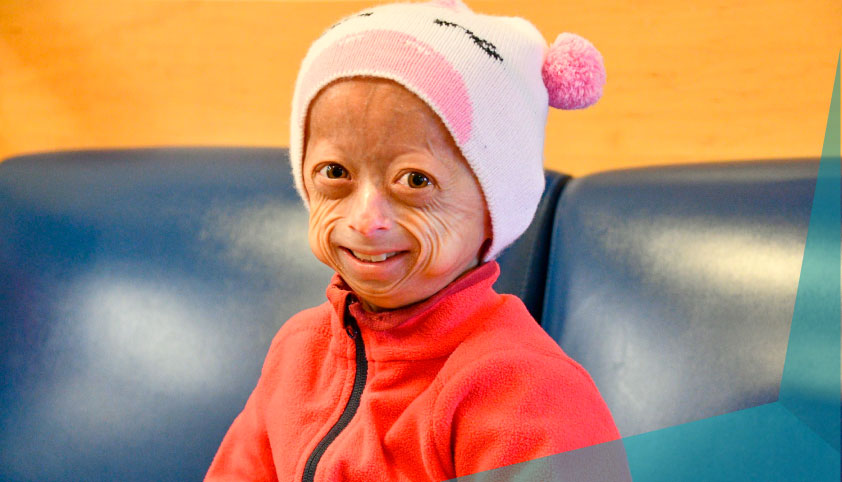
Clinical Manifestations and Progeroid Features
Patients with processing-deficient progeroid laminopathy exhibit a spectrum of systemic features that overlap with other progeroid syndromes:
- Growth retardation
- Lipodystrophy and skin atrophy
- Skeletal abnormalities and joint stiffness
- Cardiovascular disease (arteriosclerosis, valve disease)
- Insulin resistance and metabolic syndrome
- Premature aging of hair and facial features
The condition often presents in early childhood or adolescence, progressing with age due to the cumulative effects of nuclear damage.
Diagnostic Criteria and Genetic Testing
Diagnosis is established through:
- Clinical evaluation of characteristic phenotypic features
- Genetic testing revealing heterozygous mutations in the LMNA gene
- Molecular assays to detect unprocessed prelamin A and aberrant nuclear morphology in fibroblast cultures
- Imaging studies (e.g., echocardiography, DEXA scans) for systemic involvement
Next-generation sequencing (NGS) panels focused on progeroid and laminopathy-associated genes are now routinely utilized for early detection.
Differential Diagnosis
It is essential to differentiate this condition from:
- Hutchinson-Gilford Progeria Syndrome (HGPS) – classic progeria with the c.1824C>T LMNA mutation
- Mandibuloacral Dysplasia – recessive disorder with bone involvement
- Restrictive Dermopathy – neonatal lethal laminopathy due to ZMPSTE24 mutations
Each disorder has distinct molecular signatures, onset ages, and severity profiles.
Therapeutic Approaches and Management Strategies
Targeted Molecular Therapies
- Farnesyltransferase inhibitors (FTIs), such as lonafarnib, reduce prelamin A farnesylation, improving nuclear architecture and clinical parameters.
- Antisense oligonucleotides (ASOs) targeting aberrant LMNA splicing are in development for more personalized treatment.
Symptomatic and Supportive Care
- Cardiovascular monitoring and statin therapy for early atherosclerosis
- Nutritional support to counteract growth failure
- Endocrinological management for insulin resistance and lipodystrophy
- Physiotherapy to preserve joint mobility and manage musculoskeletal pain
Long-term multidisciplinary care is essential for optimizing quality of life.
Current Research and Future Directions
Advancements in gene-editing technologies such as CRISPR/Cas9 hold promise for correcting LMNA mutations at the genomic level. Preclinical studies using patient-derived induced pluripotent stem cells (iPSCs) provide insights into laminopathy pathogenesis and therapeutic screening. The integration of omics-based biomarkers is also enhancing our understanding of disease heterogeneity and progression.
Processing-deficient progeroid laminopathy with heterozygous LMNA mutation exemplifies the complex interplay between genetic mutations, nuclear architecture, and systemic disease. While current therapies focus on mitigating symptoms and halting progression, ongoing research into molecular and genomic treatments offers hope for more definitive cures. Comprehensive genetic counseling and early intervention remain vital components of patient care in managing this debilitating disorder.