Congenital Long QT Syndrome (LQTS) is a hereditary cardiac channelopathy marked by prolonged ventricular repolarization, predisposing individuals to life-threatening ventricular arrhythmias such as torsades de pointes and sudden cardiac death. Early diagnosis and effective preventive strategies are vital for risk mitigation. This article outlines an evidence-based framework for the prevention of ventricular arrhythmia in patients with congenital LQTS, focusing on genotype-specific management, pharmacologic intervention, and device therapy.
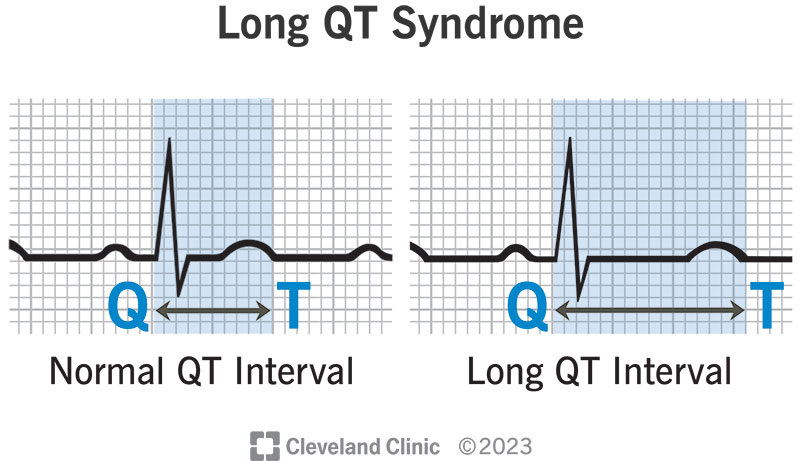
Understanding the Pathophysiology of Congenital Long QT Syndrome
Congenital LQTS is caused by mutations in genes encoding cardiac ion channels, leading to delayed ventricular repolarization. The primary subtypes include:
- LQT1 (KCNQ1 mutation): Defective slow potassium channels
- LQT2 (KCNH2 mutation): Impaired rapid potassium channels
- LQT3 (SCN5A mutation): Sodium channel dysfunction leading to prolonged depolarization
This delayed repolarization increases the QT interval on the ECG and heightens the risk for polymorphic ventricular tachycardia.
Clinical Diagnosis and Risk Stratification
Diagnostic Criteria
- QTc > 480 ms on repeated ECGs
- Schwartz Score incorporating ECG, clinical history, and family history
- Genetic confirmation via sequencing of LQTS-associated genes
Risk Stratification Factors
- QTc duration ≥ 500 ms
- Syncope or cardiac arrest history
- Male sex (during childhood), female sex (after adolescence)
- Specific genotypes (e.g., LQT2/3 carry higher event risk)
Pharmacologic Prevention: Beta-Blocker Therapy
First-Line Therapy
Beta-blockers remain the cornerstone of pharmacologic management, especially for LQT1 and LQT2. These agents reduce adrenergic stimulation, which is often the precipitating factor for arrhythmia in LQTS patients.
Recommended Beta-Blockers:
- Nadolol (40–80 mg/day): Long half-life, superior efficacy
- Propranolol (2–3 mg/kg/day): Particularly effective in pediatric cases
Genotype-Specific Efficacy
| Genotype | Beta-Blocker Effectiveness | Notes |
|---|---|---|
| LQT1 | High | Swimming is a common trigger |
| LQT2 | Moderate | Sudden auditory stimuli often involved |
| LQT3 | Limited | Bradycardia-triggered events common |
Non-Pharmacologic Measures and Lifestyle Adjustments
Activity Modification
- Avoid competitive sports in high-risk patients unless cleared by electrophysiology
- LQT1: Avoid swimming or intense exertion without proper beta-blocker coverage
- LQT2: Avoid sudden loud noises and auditory alarms
- LQT3: Avoid bradycardia-inducing environments such as deep sleep without monitoring
Medication Avoidance
Discontinue or avoid QT-prolonging drugs listed on crediblemeds.org. These include:
- Macrolide antibiotics (e.g., erythromycin)
- Antipsychotics (e.g., haloperidol)
- Antiarrhythmics (e.g., sotalol)
Implantable Cardioverter-Defibrillator (ICD) Therapy
Indications for ICD in LQTS
- Survivors of cardiac arrest
- Recurrent syncope despite optimal beta-blockade
- QTc > 550 ms with family history of sudden cardiac death
Considerations
- ICDs are life-saving but associated with complications, including inappropriate shocks and device-related anxiety, especially in younger populations.
- Subcutaneous ICDs (S-ICD) may be preferred in younger patients for reduced lead-related issues.
Left Cardiac Sympathetic Denervation (LCSD)
Indications
- High-risk patients who remain symptomatic despite maximal pharmacologic therapy
- Intolerance or contraindication to beta-blockers
- Adjunct to ICD in patients with frequent shocks
LCSD involves surgical interruption of sympathetic innervation to the heart and has demonstrated efficacy in reducing arrhythmic events in high-risk LQTS patients.
Genetic Counseling and Family Screening
Given the autosomal dominant inheritance pattern, first-degree relatives must undergo:
- ECG screening
- Genetic testing if the proband’s mutation is known
Prophylactic beta-blockers or further evaluation may be warranted based on the genetic and clinical profile of family members.
Follow-Up and Monitoring Strategies
Periodic Evaluation
- Regular ECGs to monitor QT interval
- Annual or biannual Holter monitoring
- Exercise stress testing to assess response in LQT1/2
Compliance and Therapy Adjustment
- Ensure adherence to beta-blockers
- Reassess need for ICD based on evolving risk
- Monitor for side effects such as bradycardia or hypotension
Current Guidelines and Evidence-Based Recommendations
American Heart Association (AHA) / American College of Cardiology (ACC)
- Strong recommendation for lifelong beta-blocker use in LQT1/2
- ICD in high-risk or symptomatic patients
Heart Rhythm Society (HRS)
- Genetic testing in all confirmed cases and first-degree relatives
- Lifestyle modification as adjunct to therapy
Prevention of ventricular arrhythmia in congenital long QT syndrome requires a multidisciplinary and genotype-informed approach. Optimal outcomes are achieved through early identification, appropriate pharmacologic treatment with beta-blockers, lifestyle adjustments, timely ICD implantation in selected patients, and family-based genetic counseling. Regular monitoring and individualized treatment adjustments ensure continued protection against life-threatening arrhythmias in this vulnerable population.