Pemphigus vulgaris (PV) is a rare, potentially life-threatening autoimmune disorder marked by the formation of flaccid blisters and painful erosions on the skin and mucous membranes. It results from autoantibodies directed against desmoglein 3 and, in some cases, desmoglein 1 — essential adhesion molecules within the desmosomes of epithelial cells. The consequent loss of cohesion between keratinocytes, or acantholysis, leads to intraepidermal blistering.
Epidemiology and Risk Factors of Pemphigus Vulgaris
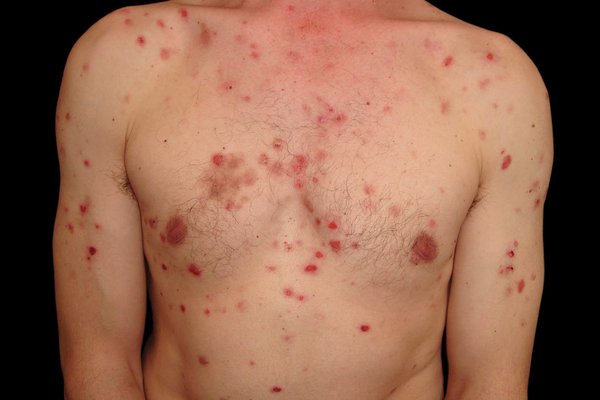
PV affects individuals globally but has a higher incidence among Ashkenazi Jews, people of Mediterranean, Middle Eastern, and South Asian descent, and typically arises between the ages of 40 and 60.
Key Risk Factors:
- Genetic predisposition: Strong association with HLA-DR4 and HLA-DR14 alleles
- Certain medications: Penicillamine, rifampin, ACE inhibitors
- Emotional or physical stress
- Viral infections or trauma
Pathogenesis of Pemphigus Vulgaris: Mechanism of Autoimmune Blistering
Pemphigus vulgaris is driven by an IgG-mediated type II hypersensitivity reaction. Autoantibodies primarily target desmoglein 3 in mucosal-dominant PV, and both desmoglein 3 and 1 in mucocutaneous types.
These pathogenic autoantibodies interrupt epithelial integrity, causing fragile blisters that rupture easily, resulting in painful denuded erosions susceptible to infection.
Clinical Features of Pemphigus Vulgaris
Mucosal Involvement
- Oral cavity is the initial site in 50–70% of cases
- Painful, non-healing ulcers affecting buccal mucosa, palate, tongue, and lips
- Can involve nasal, pharyngeal, laryngeal, conjunctival, and genital mucosa
Cutaneous Manifestations
- Flaccid bullae on normal or erythematous skin
- Lesions rupture easily, leaving moist, eroded areas with crusts
- Common sites: Scalp, face, chest, axillae, back, groin
Diagnostic Clinical Signs
- Nikolsky sign: Epidermal detachment upon gentle lateral pressure
- Asboe-Hansen sign: Blister extension into adjacent unblistered skin when pressed
Diagnostic Evaluation of Pemphigus Vulgaris
Histopathological Analysis
Biopsy of the blister reveals:
- Intraepidermal clefting
- Acantholysis with rounded keratinocytes (Tzanck cells)
- Sparing of basal layer cells (“row of tombstones” appearance)
Direct Immunofluorescence (DIF)
- Perilesional skin biopsy shows intercellular IgG and C3 deposits
- “Fishnet” or “chicken wire” pattern across epidermis confirms diagnosis
Indirect Immunofluorescence (IIF)
- Detects circulating autoantibodies in patient serum
- Performed using monkey esophagus or other epithelial substrates
ELISA Testing
- Quantifies anti-desmoglein 3 and desmoglein 1 antibodies
- Correlates with disease severity and progression
Differential Diagnoses
| Disease | Key Differences |
|---|---|
| Bullous pemphigoid | Subepidermal blister, tense bullae, elderly patients |
| Stevens-Johnson Syndrome | Drug-induced, target lesions, systemic symptoms |
| Erosive Lichen Planus | Wickham striae, no flaccid bullae |
| Mucous Membrane Pemphigoid | Scarring mucosal lesions, different immunofluorescence pattern |
Current Treatment Approaches for Pemphigus Vulgaris
First-Line Therapy: Systemic Corticosteroids
High-dose oral prednisone or prednisolone (1–1.5 mg/kg/day) remains the mainstay of initial treatment. Rapid control of inflammation and suppression of new blister formation is essential.
Steroid-Sparing Immunosuppressive Agents
- Azathioprine (2–3 mg/kg/day)
- Mycophenolate mofetil (1–2 g/day)
- Methotrexate, cyclophosphamide used in resistant cases
Biologic Therapy: Rituximab
- Anti-CD20 monoclonal antibody
- Depletes B-cells producing pathogenic antibodies
- Now FDA-approved as a first-line therapy alongside corticosteroids
- Induces durable remission in majority of patients
Adjunct Therapies
- Intravenous Immunoglobulin (IVIG): Used in severe or refractory PV
- Plasmapheresis or immunoadsorption: Helps remove circulating antibodies
- Topical corticosteroids: For localized mucosal lesions
- Antimicrobials: For secondary infection control
Monitoring and Long-Term Management
Disease Activity Assessment
- Monitor anti-desmoglein antibody titers
- Regular clinical evaluation of new erosions, mucosal involvement
- Periodic CBC, liver and renal function tests for immunosuppressive therapy side effects
Relapse Management
- Gradual tapering of corticosteroids after sustained remission
- Rituximab re-administration if relapse occurs
- Close patient education on symptom recognition
Prognosis and Quality of Life Considerations
With modern therapeutic strategies, mortality has decreased to <10%, mostly in untreated or severely immunosuppressed patients. However, pemphigus vulgaris remains a chronic disease with frequent relapses.
- Early diagnosis and aggressive initial control correlate with better outcomes
- Psychosocial support essential due to visible lesions and long-term therapy
- Multidisciplinary care (dermatology, dentistry, ophthalmology, psychology) improves patient outcomes
Preventive Measures and Patient Education
- Avoid known triggers, especially specific medications
- Emphasize adherence to therapy and follow-up schedules
- Maintain oral hygiene and nutritional support in cases with mucosal involvement
- Educate on signs of relapse and potential infections
Pemphigus vulgaris represents one of the most complex autoimmune dermatologic diseases, necessitating prompt recognition, accurate diagnosis, and comprehensive immunosuppressive therapy. With the advent of targeted biologics like rituximab, the treatment landscape has significantly evolved, offering improved remission rates and quality of life for affected individuals. Ongoing patient monitoring, education, and support are integral to successful long-term management.