Oligodendroglioma is a rare, slow-growing primary brain tumor originating from oligodendrocytes—the glial cells responsible for producing myelin in the central nervous system. These tumors are classified under diffuse gliomas, accounting for approximately 5–15% of all gliomas, and predominantly affect adults between the ages of 30 and 50.
The World Health Organization (WHO) recognizes two grades of oligodendroglioma:
- Grade 2 (low-grade): Slow-growing, less aggressive
- Grade 3 (anaplastic): Faster-growing and more aggressive
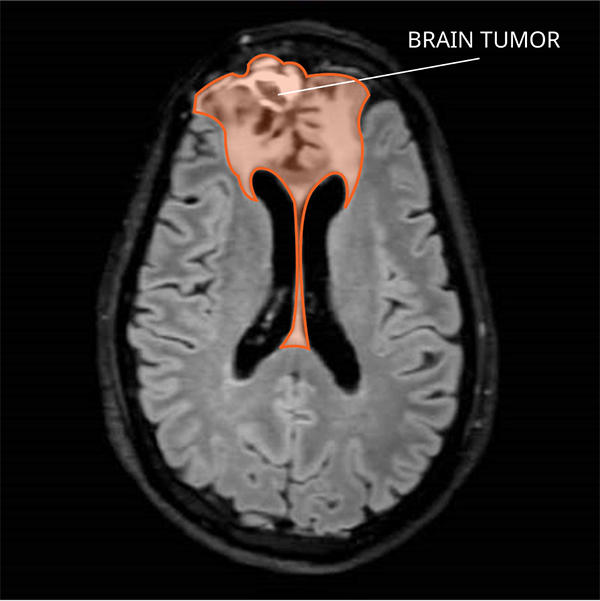
Classification of Oligodendroglioma
All oligodendrogliomas share two defining molecular characteristics:
- IDH1 or IDH2 gene mutation
- 1p/19q chromosomal codeletion
These markers are essential for accurate diagnosis and significantly influence prognosis and treatment response.
Causes and Risk Factors
The exact cause of oligodendroglioma remains unknown, though several genetic and environmental factors are associated with its development.
Genetic Markers
- IDH mutations (especially IDH1) are present in over 90% of oligodendrogliomas.
- 1p/19q codeletion, the combined loss of short arm of chromosome 1 and long arm of chromosome 19, is a hallmark of true oligodendrogliomas and correlates with better therapy responsiveness and survival rates.
Risk Factors
- Age: Commonly diagnosed between 30–50 years
- Sex: Slight male predominance
- Radiation exposure: Previous cranial irradiation may contribute
- Family history: Rare familial glioma syndromes may increase risk
Symptoms of Oligodendroglioma
Symptoms depend on tumor size, location, and growth rate. Most oligodendrogliomas occur in the frontal lobe, leading to personality or cognitive changes.
Common Clinical Features:
- Seizures (most frequent symptom)
- Headaches, often due to increased intracranial pressure
- Cognitive decline or personality changes
- Motor weakness or sensory disturbances
- Visual disturbances, if located near optic pathways
- Speech difficulties, especially in left frontal lesions
Due to their slow growth, symptoms may progress gradually over months or years.
Diagnosis of Oligodendroglioma
A combination of neurological evaluation, neuroimaging, and histopathological testing confirms the diagnosis.
Neuroimaging
- MRI (Magnetic Resonance Imaging): Preferred imaging modality; reveals a well-defined mass, often with calcifications.
- CT scan: Detects calcification; useful in emergency settings.
Biopsy and Histopathology
Surgical biopsy provides definitive diagnosis, including:
- Cell morphology: “Fried egg” appearance under microscopy
- Molecular testing: Mandatory to confirm IDH mutation and 1p/19q codeletion
Molecular Diagnostics
- IDH1/IDH2 mutation analysis
- 1p/19q fluorescence in situ hybridization (FISH)
- MGMT promoter methylation (may influence chemotherapy response)
Treatment Options for Oligodendroglioma
Treatment is tailored based on tumor grade, size, location, patient age, and overall health. A multidisciplinary approach offers the best outcomes.
1. Surgical Resection
- Maximal safe resection is the first-line therapy.
- Extent of resection directly correlates with improved progression-free and overall survival.
2. Radiation Therapy
- Standard for Grade 3 tumors or residual/recurrent Grade 2 lesions
- Helps delay tumor progression
- Modern techniques include IMRT and proton beam therapy
3. Chemotherapy
- PCV regimen (Procarbazine, Lomustine/CCNU, and Vincristine) is the traditional choice for 1p/19q-codeleted tumors.
- Temozolomide is often used as an alternative due to ease of administration and lower toxicity.
4. Watchful Waiting
- In select low-grade cases with minimal symptoms and complete resection, active surveillance may be chosen initially.
Prognosis and Survival Rates
Oligodendrogliomas are among the most treatable gliomas, especially when they harbor the 1p/19q codeletion and IDH mutation. Long-term survival is achievable with proper management.
Prognostic Indicators
| Factor | Influence |
|---|---|
| IDH mutation & 1p/19q codeletion | Improved survival |
| Younger age at diagnosis | Better outcomes |
| Extent of resection | Strong predictor of survival |
| Tumor grade | Grade 2 has better prognosis than Grade 3 |
| Performance status | Lower scores predict poorer outcomes |
Median survival:
- Grade 2: 12–15 years
- Grade 3: 6–10 years
Risk of Recurrence
Despite optimal treatment, oligodendrogliomas tend to recur, often progressing in grade. Routine monitoring with MRI scans is essential post-treatment.
Monitoring Schedule:
- Every 3–6 months for the first 2 years
- Every 6–12 months thereafter
- Prompt imaging if new neurological symptoms develop
Living with Oligodendroglioma
Management extends beyond medical intervention. Patients benefit from psychological support, rehabilitation, and lifestyle modifications.
Supportive Measures:
- Neurocognitive rehabilitation
- Seizure management
- Physical therapy for motor function preservation
- Nutritional counseling
- Psychosocial support and patient advocacy groups
Frequently Asked Questions:
Q1: Is oligodendroglioma a benign tumor?
No. Although slow-growing, it is still malignant due to its potential to infiltrate brain tissue and recur.
Q2: How is oligodendroglioma different from astrocytoma?
They arise from different glial cells and have distinct genetic profiles. Oligodendrogliomas are defined by 1p/19q codeletion and IDH mutation.
Q3: Can oligodendroglioma be cured?
While long-term control is achievable, a definitive cure is rare due to recurrence risk.
Q4: Is chemotherapy always necessary for Grade 2 tumors?
Not always. In some low-risk cases with complete resection, observation may be preferred initially.
Q5: What is the role of genetics in oligodendroglioma prognosis?
IDH mutation and 1p/19q codeletion are strong predictors of favorable response to therapy and prolonged survival.
Oligodendroglioma represents a unique subtype of brain tumors characterized by its genetic profile and relatively favorable prognosis. Prompt diagnosis, accurate molecular classification, and an individualized treatment plan are critical for optimizing outcomes. Long-term surveillance and holistic care remain the cornerstone of managing this chronic neurological condition.