Lymphangioleiomyomatosis (LAM) is a rare, progressive lung disease primarily affecting women of childbearing age. It is characterized by the abnormal proliferation of smooth muscle-like cells (LAM cells) in the lungs, leading to cystic destruction, airflow obstruction, and eventual respiratory failure. LAM can occur sporadically or in association with Tuberous Sclerosis Complex (TSC-LAM).
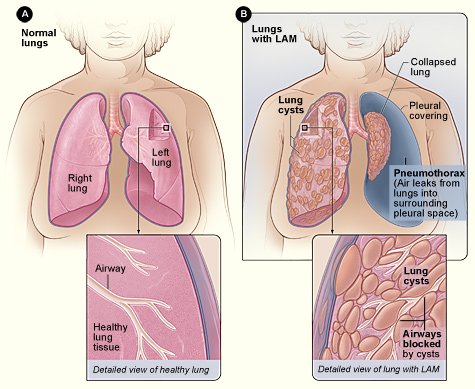
Pathophysiology of LAM
LAM cells originate from mutations in the TSC1 or TSC2 genes, which regulate the mTOR (mammalian target of rapamycin) pathway. Dysfunction in this pathway leads to uncontrolled cell growth, cyst formation, and lung damage.
Causes and Risk Factors
The exact cause of sporadic LAM remains unknown, but the following factors contribute to disease development:
- Genetic Mutations – TSC1 or TSC2 mutations result in abnormal cellular proliferation.
- Hormonal Influence – LAM worsens during pregnancy and is rare in men, indicating an estrogen-dependent mechanism.
- Tuberous Sclerosis Complex (TSC) – Women with TSC are at higher risk of developing LAM.
Signs and Symptoms of LAM
LAM symptoms vary in severity and progression. Common clinical manifestations include:
- Progressive Dyspnea (Shortness of Breath) – Due to airflow obstruction and lung damage.
- Recurrent Pneumothorax (Collapsed Lung) – Cyst rupture can cause air leakage into the pleural cavity.
- Chylous Pleural Effusion – Lymphatic obstruction leads to fluid accumulation in the pleural space.
- Chronic Cough – Persistent, often non-productive cough.
- Hemoptysis (Coughing up Blood) – Caused by fragile blood vessels in the lungs.
- Fatigue and Chest Pain – Common in advanced cases.
Diagnosis of LAM
1. High-Resolution Computed Tomography (HRCT) Scan
HRCT is the gold standard for LAM diagnosis, showing characteristic thin-walled lung cysts distributed throughout both lungs.
2. Pulmonary Function Tests (PFTs)
PFTs assess lung capacity and airflow limitation, revealing:
✔ Reduced Forced Expiratory Volume (FEV1)
✔ Increased Residual Volume (RV) due to air trapping
3. Serum Vascular Endothelial Growth Factor-D (VEGF-D) Test
Elevated VEGF-D levels (>800 pg/mL) strongly indicate LAM, reducing the need for lung biopsy.
4. Lung Biopsy
In uncertain cases, transbronchial lung biopsy or video-assisted thoracoscopic surgery (VATS) biopsy confirms LAM by detecting HMB-45-positive LAM cells.
5. Genetic Testing
TSC1/TSC2 gene mutation analysis is recommended in suspected TSC-LAM cases.
Treatment Options for Lymphangioleiomyomatosis
1. mTOR Inhibitors (Sirolimus, Everolimus)
Sirolimus (Rapamycin) is the first-line therapy for LAM, effectively stabilizing lung function by inhibiting the mTOR pathway. Benefits include:
✔ Slowing lung function decline
✔ Reducing VEGF-D levels
✔ Decreasing chylous effusion recurrence
2. Oxygen Therapy
Supplemental oxygen is required in advanced LAM cases with severe hypoxemia to improve quality of life.
3. Bronchodilators
Short-acting beta-agonists (e.g., Albuterol) help manage airflow obstruction in LAM patients with reversible airway involvement.
4. Pleurodesis for Pneumothorax
Recurrent pneumothorax is common in LAM. Chemical (talc) or mechanical pleurodesis prevents lung collapse by fusing the pleural layers.
5. Lung Transplantation
For end-stage LAM, lung transplantation remains the only curative option. Survival rates after transplantation exceed 50% at five years.
Prognosis and Life Expectancy
LAM progression varies among individuals. Median survival post-diagnosis is 20-30 years, with sirolimus significantly improving outcomes. Factors influencing prognosis include:
- Rate of lung function decline (FEV1 decline >100 mL/year worsens prognosis)
- VEGF-D levels (Higher levels correlate with severe disease)
- Response to mTOR inhibitors
Future Research and Emerging Therapies
1. Gene Therapy Approaches
CRISPR-based TSC1/TSC2 gene editing is under investigation to correct LAM-associated mutations.
2. VEGF-D Targeted Therapy
New monoclonal antibodies against VEGF-D may provide alternative treatment strategies.
3. Combination Therapy
Research is exploring sirolimus plus autophagy inhibitors to enhance treatment efficacy.
Frequently Asked Questions:
1. Is Lymphangioleiomyomatosis Cancerous?
No, LAM is not cancer, but LAM cells share similarities with tumor cells, leading to invasive growth in the lungs.
2. Can LAM Be Cured?
There is no cure, but treatments like Sirolimus slow disease progression and improve quality of life.
3. How Fast Does LAM Progress?
LAM progression varies; some patients experience slow decline, while others develop rapid lung deterioration.
4. Is LAM Hereditary?
Sporadic LAM is not inherited, but TSC-LAM is genetic, occurring in TSC patients due to TSC1/TSC2 mutations.
5. Can Men Get LAM?
LAM is extremely rare in men, further supporting its hormone-dependent nature.