Homozygous familial hypercholesterolemia (HoFH) is a rare but severe genetic disorder that results in extremely high levels of low-density lipoprotein cholesterol (LDL-C) from birth. Without prompt and aggressive treatment, HoFH can lead to early cardiovascular disease and reduced life expectancy.
What is Homozygous Familial Hypercholesterolemia?
HoFH is an inherited condition where a person receives defective LDL receptor genes from both parents. This results in impaired clearance of LDL cholesterol from the bloodstream, leading to cholesterol levels several times higher than normal. Unlike heterozygous familial hypercholesterolemia (HeFH), which presents milder symptoms, HoFH is far more aggressive and requires intensive management.
Causes of Homozygous Familial Hypercholesterolemia
HoFH is caused by mutations in the LDL receptor (LDLR) gene, the apolipoprotein B (APOB) gene, or the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene. The absence or dysfunction of LDL receptors significantly impairs the body’s ability to remove LDL cholesterol from the bloodstream.
Symptoms of Homozygous Familial Hypercholesterolemia
Common symptoms of HoFH include:
- Extremely elevated LDL cholesterol levels (often above 500 mg/dL)
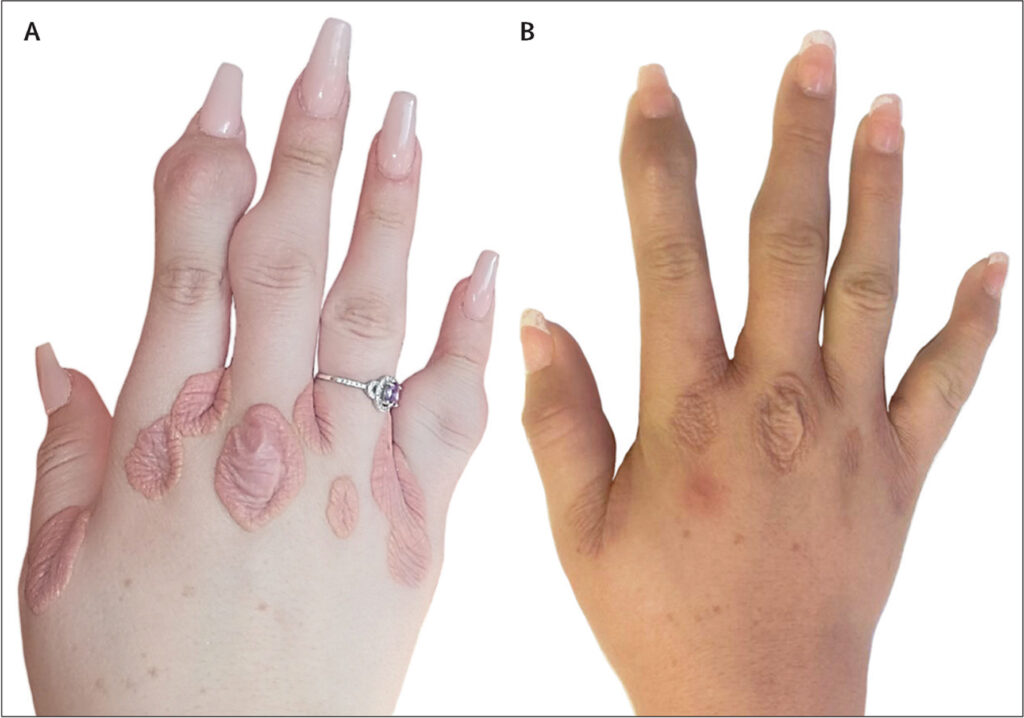
- Xanthomas (fatty deposits) on the skin, particularly on elbows, knees, and buttocks
- Xanthelasmas (yellowish deposits) around the eyes
- Corneal arcus (a white or gray ring around the cornea)
- Early-onset cardiovascular disease (before age 20 in severe cases)
Diagnosis of Homozygous Familial Hypercholesterolemia
Diagnostic steps typically include:
- Lipid Profile Test: Identifying LDL cholesterol levels exceeding 500 mg/dL
- Genetic Testing: Identifying mutations in LDLR, APOB, or PCSK9 genes
- Family History Assessment: Understanding familial patterns of cholesterol disorders
- Physical Examination: Identifying xanthomas, xanthelasmas, or corneal arcus
Treatment for Homozygous Familial Hypercholesterolemia
Effective management of HoFH requires a combination of aggressive therapies:
1. Medications
- Statins: Reduce cholesterol production in the liver
- Ezetimibe: Limits intestinal cholesterol absorption
- PCSK9 Inhibitors: Enhance LDL receptor recycling for better cholesterol clearance
- Mipomersen and Lomitapide: Approved for severe HoFH cases to reduce LDL cholesterol
2. Lipoprotein Apheresis
A dialysis-like procedure that physically removes LDL cholesterol from the bloodstream. This is often necessary for individuals whose cholesterol levels remain dangerously high despite medication.
3. Liver Transplant
In extreme cases where other treatments fail, liver transplantation can improve LDL receptor function, significantly lowering cholesterol levels.
4. Diet and Lifestyle Changes
- Adopt a low-fat, heart-healthy diet
- Increase physical activity
- Avoid smoking and limit alcohol intake
Potential Complications
Without appropriate management, HoFH may lead to:
- Premature atherosclerosis
- Heart attacks in childhood or adolescence
- Stroke or other cardiovascular emergencies
Prognosis and Life Expectancy
With early diagnosis and aggressive treatment, individuals with HoFH can significantly improve their life expectancy. Lifelong adherence to medical therapies, combined with regular monitoring, is essential for effective management.
Genetic Counseling and Family Screening
Since HoFH is an inherited condition, genetic counseling is crucial for affected families. Early screening of siblings and other family members helps in timely diagnosis and treatment.
Homozygous familial hypercholesterolemia is a life-threatening genetic disorder that demands urgent and sustained intervention. With advancements in medical treatments and awareness, those diagnosed with HoFH can achieve improved health outcomes and enhanced quality of life.