Hemophagocytic lymphohistiocytosis (HLH) is a rare but potentially life-threatening immune disorder characterized by excessive immune activation. It leads to severe inflammation and multi-organ dysfunction. HLH can be classified into primary (genetic) and secondary (acquired) forms, each presenting distinct challenges in diagnosis and treatment.
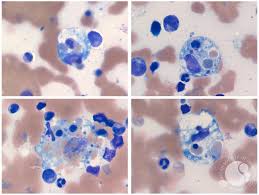
Types of Hemophagocytic Lymphohistiocytosis
Primary HLH (Familial HLH)
Primary HLH is an inherited condition caused by genetic mutations affecting immune cell function. It typically manifests in infancy or early childhood and requires genetic testing for confirmation.
Secondary HLH (Acquired HLH)
Secondary HLH is triggered by infections, autoimmune diseases, malignancies, or immunosuppressive therapies. It can occur at any age and is often associated with severe underlying conditions.
Causes and Risk Factors
Genetic Causes
- Mutations in genes like PRF1, UNC13D, STXBP2, and RAB27A.
- Autosomal recessive inheritance patterns in most primary HLH cases.
Acquired Triggers
- Viral Infections: Epstein-Barr virus (EBV), cytomegalovirus (CMV), or HIV.
- Bacterial Infections: Tuberculosis, brucellosis, or sepsis.
- Autoimmune Conditions: Systemic lupus erythematosus (SLE), rheumatoid arthritis.
- Malignancies: Lymphoma, leukemia.
Symptoms of Hemophagocytic Lymphohistiocytosis
HLH symptoms are often non-specific and can mimic severe systemic inflammation or infection. Common symptoms include:
- Prolonged fever exceeding seven days.
- Enlarged liver and spleen (hepatosplenomegaly).
- Pancytopenia (reduced blood cell counts).
- Skin rashes and lymph node swelling.
- Neurological symptoms like seizures, irritability, or altered mental status.
Diagnostic Criteria for HLH
The diagnosis of HLH requires meeting five of the following eight criteria:
- Fever ≥38°C.
- Splenomegaly.
- Cytopenias affecting at least two cell lines.
- Elevated ferritin levels.
- Elevated soluble CD25 (sIL-2R).
- Hemophagocytosis observed in bone marrow, spleen, or lymph nodes.
- Low or absent NK cell activity.
- Elevated triglycerides or low fibrinogen.
Treatment of Hemophagocytic Lymphohistiocytosis
Initial Therapy
- Dexamethasone: A key corticosteroid used to reduce inflammation.
- Etoposide: A chemotherapy agent effective in controlling HLH symptoms.
- Intravenous immunoglobulin (IVIG): Helps regulate the immune response.
- Cyclosporine A: Used for immune modulation.
Advanced Treatment Options
- Hematopoietic stem cell transplantation (HSCT): Recommended for primary HLH patients with persistent disease or relapse.
- Biologic therapies such as emapalumab target interferon-gamma pathways to suppress excessive inflammation.
Prognosis and Survival Rates
Outcomes depend on early diagnosis and appropriate treatment. With prompt intervention, survival rates in primary HLH patients have improved significantly. For secondary HLH, successful treatment of the underlying condition is critical to improving prognosis.
Preventive Measures and Management
- Genetic counseling for families with a history of primary HLH.
- Regular monitoring for patients with autoimmune conditions or malignancies.
- Prophylactic antiviral or antibiotic therapy in immunocompromised patients.
Frequently Asked Questions:
What is the most common cause of secondary HLH?
Infections, particularly Epstein-Barr virus (EBV), are the most common triggers of secondary HLH.
Can HLH be cured?
While secondary HLH can often be resolved by treating the underlying condition, primary HLH requires aggressive therapy and may necessitate stem cell transplantation.
How long does HLH treatment last?
Treatment duration varies; induction therapy typically lasts eight weeks, while maintenance treatment can continue for several months.
What are the risks of untreated HLH?
Untreated HLH can lead to severe organ damage, multi-organ failure, and death.
Is HLH contagious?
HLH itself is not contagious, but certain viral triggers such as EBV may be transmitted between individuals.
Hemophagocytic lymphohistiocytosis is a complex and potentially fatal condition requiring timely diagnosis and aggressive treatment. Understanding its causes, symptoms, and management strategies is crucial for improving patient outcomes.