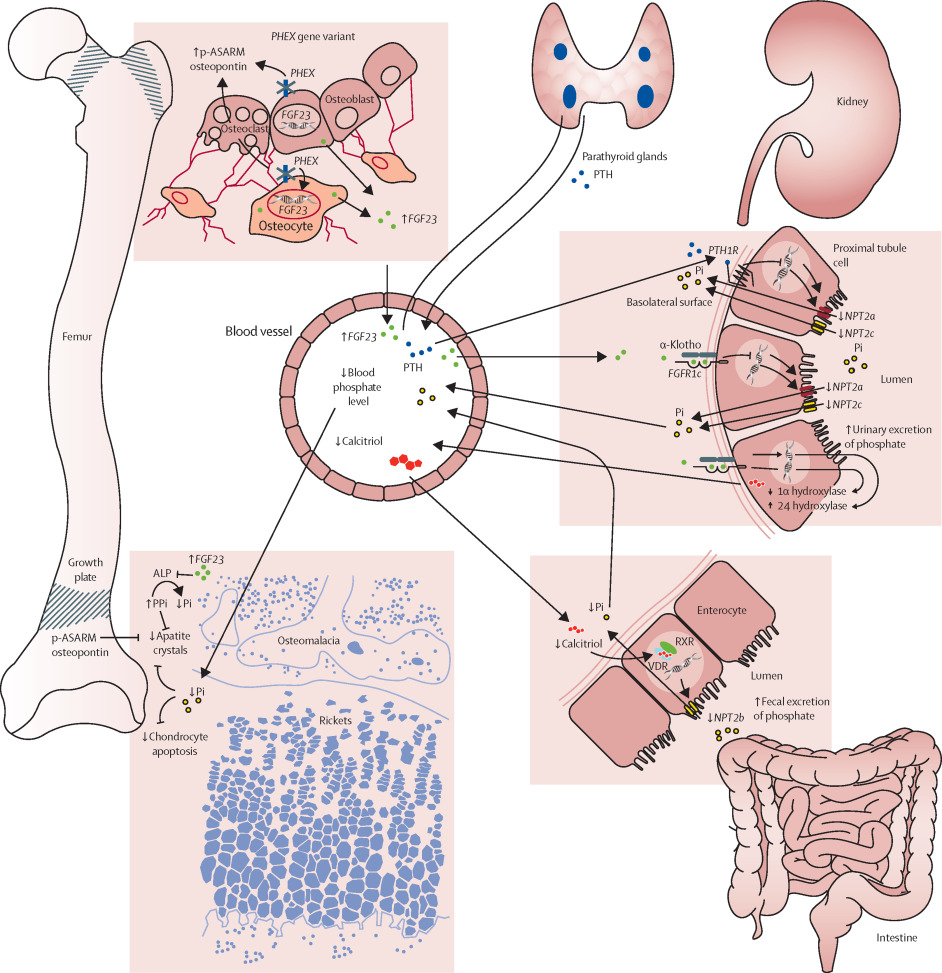
X-linked hypophosphatemic osteomalacia (XLH) is a rare, inherited phosphate-wasting disorder that affects bone mineralization. It is the most common genetic form of rickets and osteomalacia, resulting from mutations in the PHEX gene located on the X chromosome. This mutation leads to increased levels of fibroblast growth factor 23 (FGF23), which inhibits renal phosphate reabsorption and suppresses vitamin D synthesis, ultimately causing hypophosphatemia and impaired bone formation.
Genetic Basis and Inheritance Pattern
XLH follows an X-linked dominant inheritance, meaning it affects both males and females. However, male patients often exhibit more severe phenotypes. The PHEX gene mutation leads to dysregulation of FGF23, resulting in phosphate loss through urine and inadequate activation of vitamin D.
Pathophysiology: Phosphate Wasting and Impaired Bone Mineralization
In healthy individuals, phosphate is reabsorbed in the renal tubules and supports bone mineralization. In XLH:
- PHEX mutations impair the breakdown of FGF23
- FGF23 excess suppresses sodium-phosphate cotransporters in renal tubules
- Reduced phosphate reabsorption leads to hypophosphatemia
- Impaired 1-α-hydroxylation of 25-hydroxyvitamin D results in low calcitriol (active vitamin D)
- Poor calcium-phosphate deposition in bones causes osteomalacia
Clinical Features of X-Linked Hypophosphatemic Osteomalacia
Pediatric Presentation (Rickets)
- Short stature due to stunted bone growth
- Bowing of the legs (genu varum)
- Delayed walking or waddling gait
- Dental abscesses without caries due to defective dentin
- Craniosynostosis in severe cases
Adult Manifestations (Osteomalacia)
- Bone pain and tenderness
- Muscle weakness
- Fatigue
- Fractures or pseudofractures, particularly in the femur and tibia
- Enthesopathy, including calcifications at tendon insertions
- Spinal stenosis or joint deformities in chronic cases
Diagnostic Approach to XLH
1. Laboratory Investigations
- Low serum phosphate
- Normal or elevated PTH
- Normal calcium
- Low or inappropriately normal 1,25-dihydroxyvitamin D
- Elevated alkaline phosphatase in children
- Elevated FGF23 levels (in untreated cases)
2. Radiographic Findings
- Widened, irregular growth plates
- Cupping and fraying of metaphyses
- Looser zones or pseudofractures in adult bones
3. Genetic Testing
- Detection of mutations in the PHEX gene confirms the diagnosis
- May also be used for prenatal screening or carrier detection
Differential Diagnosis of Hypophosphatemic Osteomalacia
| Condition | Distinguishing Features |
|---|---|
| Tumor-induced osteomalacia | Acquired, FGF23-secreting tumor, usually in adults |
| Autosomal dominant hypophosphatemic rickets | Mutation in FGF23 gene |
| Hereditary hypophosphatemic rickets with hypercalciuria | Mutation in SLC34A3; hypercalciuria present |
| Nutritional rickets | Low calcium, low vitamin D intake, not hereditary |
Treatment Strategies for XLH
1. Conventional Therapy
- Oral phosphate supplements (multiple doses daily)
- Active vitamin D analogs (calcitriol or alfacalcidol)
- Monitoring for:
- Hyperparathyroidism
- Nephrocalcinosis
- Secondary calcium imbalances
2. Burosumab Therapy (Anti-FGF23 Monoclonal Antibody)
- FDA-approved biologic for XLH treatment in both children and adults
- Binds to and inhibits excess FGF23
- Normalizes phosphate levels and improves:
- Bone mineralization
- Mobility
- Fracture healing
- Administered via subcutaneous injection every 2–4 weeks
3. Surgical Correction
- Reserved for severe skeletal deformities unresponsive to medical treatment
- Osteotomies may be performed to correct limb bowing or joint misalignment
Long-Term Management and Monitoring
- Regular growth assessments in children
- Bone mineral density scans (DEXA)
- Renal ultrasound to check for nephrocalcinosis
- Dental evaluations to prevent or treat dental abscesses
- Physical therapy to maintain muscle strength and mobility
Psychosocial and Quality of Life Considerations
Patients with XLH often face challenges including:
- Chronic pain and fatigue
- Social limitations due to skeletal deformities
- Reduced physical performance
- Psychological impacts in adolescents and adults
Multidisciplinary care involving endocrinologists, nephrologists, orthopedists, dentists, and physical therapists is critical for comprehensive management.
Emerging Research and Future Directions
1. Gene Therapy
- Investigational therapies aim to correct PHEX mutations
- Early studies focus on viral vector delivery systems targeting bone cells
2. Next-Generation Biologics
- Research continues into more targeted FGF23 inhibitors
- Exploration of combination therapies to enhance skeletal outcomes
3. Biomarker Development
- Identification of more precise markers to monitor treatment response and progression
Frequently Asked Questions:
Q1: Is XLH curable?
No. XLH is a chronic condition, but early and appropriate treatment significantly improves outcomes.
Q2: Can females have symptoms of XLH?
Yes. XLH is X-linked dominant, so heterozygous females often show symptoms, though typically milder than males.
Q3: What is the role of FGF23 in XLH?
FGF23 reduces phosphate reabsorption and vitamin D activation; excess levels in XLH lead to hypophosphatemia.
Q4: When should burosumab be considered?
Burosumab is recommended for patients with persistent symptoms or poor response to conventional therapy.
Q5: Are there dietary recommendations for XLH?
A balanced diet with adequate phosphate and calcium is essential; however, dietary changes alone are not sufficient treatment.
X-linked hypophosphatemic osteomalacia is a genetically driven phosphate metabolism disorder with lifelong implications. With advances in targeted therapies such as burosumab and ongoing research into gene correction, the outlook for patients continues to improve. Early diagnosis, individualized treatment plans, and comprehensive multidisciplinary care are essential for minimizing complications and optimizing quality of life.