X-linked agammaglobulinemia (XLA), also known as Bruton’s agammaglobulinemia, is a rare primary immunodeficiency characterized by an almost complete absence of mature B lymphocytes and immunoglobulins in the blood. Caused by mutations in the BTK (Bruton Tyrosine Kinase) gene, this condition impairs the body’s ability to produce antibodies, rendering affected individuals highly susceptible to recurrent bacterial infections from infancy.
Genetic Mechanism and Inheritance Pattern
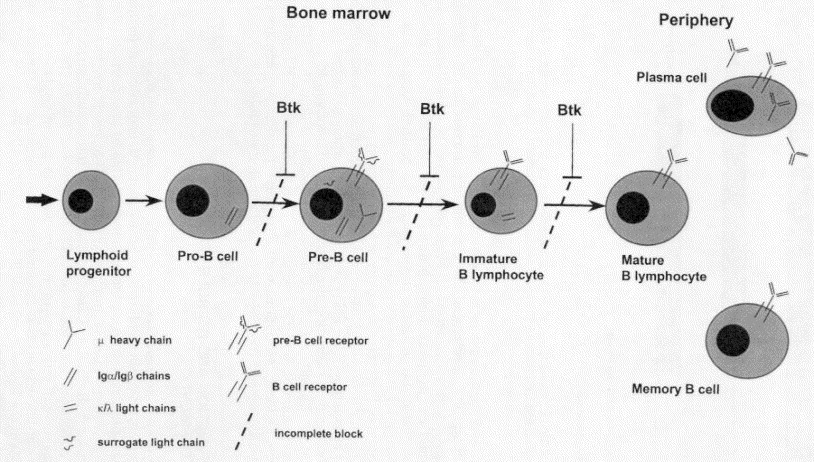
XLA is inherited in an X-linked recessive pattern, primarily affecting males. The BTK gene, located on the X chromosome at Xq21.3-Xq22, is essential for B cell development. Mutations in this gene result in an arrest of B cell maturation at the pre-B cell stage.
Pathophysiology: B Cell Arrest and Antibody Deficiency
Bruton Tyrosine Kinase (BTK) plays a pivotal role in B cell receptor signaling. A deficiency or malfunction of BTK disrupts intracellular signaling required for B cell maturation, leading to:
- Absence of mature B cells in peripheral blood and lymphoid tissues
- Deficiency of all major immunoglobulin classes (IgG, IgA, IgM, IgE)
- Compromised humoral immune response, making patients prone to infections
Clinical Manifestations of X-Linked Agammaglobulinemia
XLA typically presents in male infants after maternal IgG levels wane, usually around 6 months of age. Key clinical features include:
1. Recurrent Bacterial Infections
- Otitis media, sinusitis, bronchitis, and pneumonia caused by Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus
- Gastrointestinal infections, especially Giardia lamblia
- Skin and soft tissue infections
2. Poor Response to Vaccinations
- Ineffective immune response to both live and inactivated vaccines due to lack of antibody production
3. Absence of Lymphoid Tissue
- Tonsils, adenoids, and peripheral lymph nodes are often underdeveloped or absent
4. Chronic Lung Disease
- Untreated or recurrent respiratory infections may lead to bronchiectasis and chronic lung damage
Diagnostic Approach for XLA
Early and accurate diagnosis of XLA is crucial to prevent irreversible complications. The following tests are routinely employed:
1. Serum Immunoglobulin Levels
- Markedly reduced or absent levels of IgG, IgA, IgM, and IgE
2. Flow Cytometry
- Absence or severe reduction in CD19+ and CD20+ B lymphocytes
3. BTK Gene Analysis
- Confirmatory genetic testing to detect mutations in the BTK gene
4. Newborn Screening
- Emerging programs include severe antibody deficiencies in neonatal panels through TREC/KREC analysis
Differential Diagnosis: Distinguishing XLA from Similar Disorders
Differentiation from other primary immunodeficiencies is essential for proper management. XLA must be distinguished from:
| Condition | Distinguishing Features |
|---|---|
| Common Variable Immunodeficiency (CVID) | Later onset, presence of B cells, variable Ig levels |
| Hyper-IgM Syndrome | Elevated or normal IgM, T cell defects |
| Transient Hypogammaglobulinemia of Infancy | Self-limited, normal B cell numbers |
| Selective IgA Deficiency | Isolated low IgA, often asymptomatic |
Management and Treatment Strategies for XLA
1. Immunoglobulin Replacement Therapy
- Intravenous (IVIG) or Subcutaneous Immunoglobulin (SCIG)
- Administered every 3–4 weeks (IVIG) or weekly (SCIG)
- Maintains protective IgG levels and prevents infections
2. Antibiotic Therapy
- Prompt treatment of infections with broad-spectrum antibiotics
- Prophylactic antibiotics may be prescribed in recurrent cases
3. Live Vaccines Contraindication
- Live attenuated vaccines (e.g., MMR, Varicella, OPV) are strictly avoided due to risk of severe disease
4. Pulmonary Care
- Routine imaging and lung function tests
- Chest physiotherapy for patients with bronchiectasis
5. Genetic Counseling
- Important for families with a known mutation to assess carrier status and guide future pregnancies
Long-Term Prognosis and Quality of Life
With early diagnosis and consistent immunoglobulin therapy, individuals with XLA can lead relatively healthy lives. However, challenges may include:
- Chronic respiratory complications in poorly managed cases
- Increased risk of autoimmune manifestations and enteroviral infections
- Rare association with malignancies such as lymphoma
Regular follow-up with immunology specialists, adherence to therapy, and infection vigilance are essential for optimal outcomes.
Research and Advances in XLA Therapy
1. Gene Therapy
- Experimental treatments aim to correct the BTK mutation using viral vectors or CRISPR-based approaches
2. BTK Inhibitor Studies
- Investigating selective BTK modulators that may restore partial B cell function or prevent disease progression
3. Novel Immunoglobulin Formulations
- Long-acting IG therapies under development to reduce infusion frequency
Frequently Asked Questions:
Q1: Can XLA be cured?
Currently, XLA has no cure. Lifelong immunoglobulin replacement therapy is the standard of care.
Q2: How is XLA inherited?
XLA is inherited in an X-linked recessive manner and primarily affects males born to carrier mothers.
Q3: Can females get XLA?
Females are typically carriers; rare symptomatic cases may occur due to skewed X-inactivation.
Q4: What age does XLA present?
Symptoms usually appear after 6 months of age, once maternal antibodies decline.
Q5: Is XLA the same as CVID?
No, XLA presents in infancy with no mature B cells, while CVID has a later onset and retains B cells with defective function.
X-linked agammaglobulinemia is a serious but manageable primary immunodeficiency. Through vigilant monitoring, regular immunoglobulin therapy, and avoidance of live vaccines, most patients can enjoy a significantly improved life expectancy and quality of life. Ongoing research in gene therapy and immune modulation holds the promise of transformative treatments for future generations.