Wilson’s disease is a rare autosomal recessive genetic disorder characterized by excessive accumulation of copper in vital organs, primarily the liver and brain. It is caused by mutations in the ATP7B gene, responsible for encoding a protein crucial in copper transport. In healthy individuals, copper is absorbed from food and excess amounts are excreted via bile. However, in individuals with Wilson’s disease, this excretion mechanism fails, resulting in toxic buildup.

Genetic Cause and Pathophysiology
The primary defect lies in the ATP7B gene located on chromosome 13. This mutation impairs copper-transporting ATPase, disrupting copper incorporation into ceruloplasmin and subsequent biliary excretion. As copper accumulates, it first overwhelms hepatic stores and then spills over into the bloodstream, depositing in other tissues such as the brain, kidneys, and cornea.
Clinical Manifestations of Wilson’s Disease
The clinical presentation is highly variable and age-dependent, often categorized into hepatic, neurological, and psychiatric manifestations:
Hepatic Symptoms
- Hepatitis-like symptoms (jaundice, hepatomegaly)
- Cirrhosis
- Acute liver failure in adolescents and young adults
Neurological and Psychiatric Symptoms
- Tremors, dystonia, dysarthria
- Parkinsonism-like features
- Cognitive decline, personality changes, depression
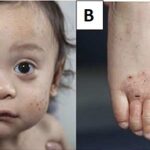
Ophthalmologic Signs
- Kayser-Fleischer Rings: Copper deposits in the Descemet’s membrane of the cornea, visible on slit-lamp examination
Diagnostic Criteria and Tests
Early diagnosis is critical to preventing irreversible damage. The diagnostic workup includes:
Laboratory Tests
- Serum ceruloplasmin: Typically low (<20 mg/dL)
- 24-hour urinary copper excretion: Elevated (>100 µg/24 hours)
- Hepatic copper quantification: >250 µg/g dry weight confirms diagnosis
Imaging
- MRI brain: Shows hyperintensities in basal ganglia and thalamus
- Liver ultrasound: May reveal fatty infiltration or cirrhotic changes
Genetic Testing
- Confirms mutations in ATP7B, particularly in familial cases
Differential Diagnosis
Due to its wide-ranging symptoms, Wilson’s disease can mimic various conditions such as:
- Autoimmune hepatitis
- Huntington’s disease
- Parkinson’s disease
- Multiple sclerosis
- Psychiatric disorders (e.g., schizophrenia, bipolar disorder)
Treatment and Long-Term Management
Copper Chelation Therapy
- D-penicillamine: First-line chelator, increases urinary copper excretion
- Trientine: Alternative for penicillamine-intolerant patients
Zinc Therapy
- Inhibits intestinal absorption of copper
- Often used for maintenance therapy
Diet Recommendations
- Avoid high-copper foods: liver, shellfish, chocolate, nuts, mushrooms
Liver Transplantation
- Considered in fulminant hepatic failure or decompensated cirrhosis unresponsive to medical therapy
Prognosis and Life Expectancy
With early diagnosis and adherence to treatment, the prognosis is favorable. Many patients live normal lives with minimal limitations. Untreated, the disease is fatal due to hepatic or neurological complications.
Wilson’s Disease in Pediatrics
Children may present with subtle hepatic signs or failure to thrive. Pediatricians should maintain a high index of suspicion, especially in cases of unexplained liver enzyme elevations or behavioral changes.
Wilson’s Disease and Pregnancy
Properly treated women with Wilson’s disease can have successful pregnancies. Preconception planning and coordination between hepatologists and obstetricians are essential.
Recent Advances and Research Directions
- Gene Therapy: Investigational approaches aim to correct the defective ATP7B gene.
- Novel Chelators: Research is ongoing into drugs with fewer side effects and better patient adherence.
- Biomarkers: Improved biomarkers for earlier diagnosis and monitoring of treatment response.
Wilson’s disease, though rare, is a serious yet treatable condition. Increased awareness, prompt diagnosis, and lifelong management are pivotal to improving outcomes. Clinicians must recognize its protean manifestations to ensure timely intervention.