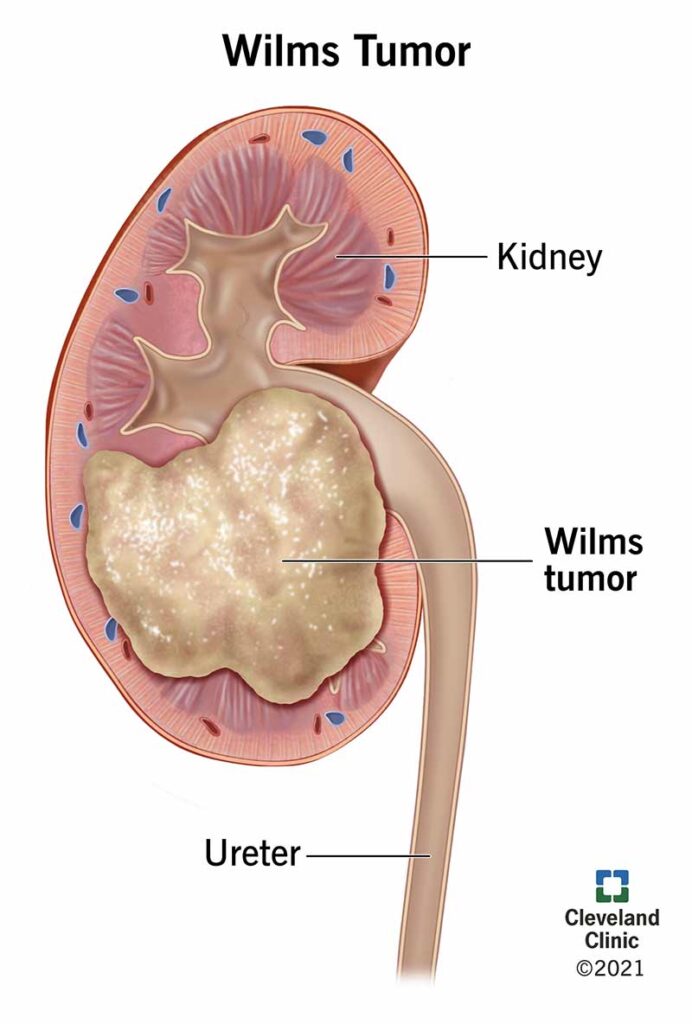
Wilms’ tumor, also known as nephroblastoma, is the most common form of kidney cancer in children. It primarily affects children under the age of five and accounts for approximately 6% of all childhood cancers. Due to advancements in diagnostic techniques and multimodal therapy, Wilms’ tumor now boasts a survival rate exceeding 90% in developed countries.
This malignant embryonal neoplasm arises from nephrogenic rests—precursors to renal tissue—that fail to differentiate appropriately during fetal development. While most cases are unilateral and sporadic, about 5–10% present bilaterally or are linked to hereditary syndromes.
Wilms’ Tumor Pathophysiology and Genetic Basis
The pathogenesis of Wilms’ tumor involves aberrant gene expression regulating renal development. The most commonly implicated genes include:
- WT1 gene (11p13): A tumor suppressor gene mutated in ~15% of cases
- WT2 locus (11p15): Associated with epigenetic changes and IGF2 overexpression
- WTX, TP53, CTNNB1, and others: Associated with tumor progression and histologic subtype
Syndromes Associated with Wilms’ Tumor:
- WAGR Syndrome (Wilms’ tumor, Aniridia, Genitourinary anomalies, and mental Retardation)
- Denys-Drash Syndrome
- Beckwith-Wiedemann Syndrome
These syndromes necessitate genetic counseling and regular surveillance to enable early detection.
Clinical Presentation and Symptoms of Wilms’ Tumor
Wilms’ tumor typically presents as a painless abdominal mass. Other symptoms include:
- Abdominal swelling or firmness
- Hematuria (blood in urine)
- Hypertension
- Fever
- Anorexia or weight loss
- Nausea or vomiting
- Constipation
Due to its asymptomatic nature in early stages, it is often discovered incidentally by parents or physicians during routine examination.
Diagnostic Evaluation and Imaging of Wilms’ Tumor
A thorough diagnostic workup is essential for accurate staging and treatment planning. The evaluation includes:
Imaging Studies
- Abdominal Ultrasound: First-line imaging to detect renal masses
- CT or MRI Scan: Detailed anatomic visualization of the tumor, contralateral kidney, and vasculature
- Chest X-ray/CT: To assess for pulmonary metastases, which are common in advanced stages
Laboratory Tests
- Complete blood count (CBC)
- Renal function tests
- Liver function tests
- Urinalysis
- Coagulation profile (especially if Denys-Drash is suspected)
Histopathological Confirmation
Diagnosis is confirmed by surgical biopsy or nephrectomy specimen. Wilms’ tumor histology is categorized as:
- Favorable histology: Classic triphasic pattern (blastemal, epithelial, stromal)
- Unfavorable histology (anaplastic): Poorer prognosis, often associated with TP53 mutations
Staging of Wilms’ Tumor
Staging is crucial for treatment planning and is based on the National Wilms Tumor Study (NWTS) system.
Treatment Modalities for Wilms’ Tumor
Treatment involves a multidisciplinary approach integrating surgery, chemotherapy, and radiotherapy, tailored according to tumor stage and histologic subtype.
Surgical Management
- Radical nephrectomy is the primary intervention for unilateral tumors.
- Nephron-sparing surgery is considered in bilateral disease or solitary kidneys.
- Lymph node sampling is essential for accurate staging.
Chemotherapy
Chemotherapy regimens are risk-adapted:
| Regimen | Drugs Included | Used For |
|---|---|---|
| EE-4A | Vincristine + Dactinomycin | Stage I–II, favorable histology |
| DD-4A | Vincristine + Dactinomycin + Doxorubicin | Stage III, IV or higher-risk cases |
| Regimen M | Additional agents (Cyclophosphamide, Etoposide) | Anaplastic histology or relapse |
Radiotherapy
- Indicated for Stage III–IV or anaplastic histology
- Radiation dose and field depend on tumor location, spread, and response to chemotherapy
Prognosis and Survival Rates
The prognosis for Wilms’ tumor has significantly improved due to optimized protocols. Survival outcomes by stage are as follows:
| Stage | 5-Year Survival Rate |
|---|---|
| Stage I | > 95% |
| Stage II | 90–95% |
| Stage III | 85–90% |
| Stage IV | 70–85% |
| Stage V | ~90% (with bilateral management) |
Factors influencing prognosis:
- Histology (favorable vs. anaplastic)
- Tumor stage at diagnosis
- Genetic mutations (e.g., TP53)
- Patient age and tumor size
Surveillance and Long-Term Follow-Up
Long-term surveillance is essential due to risks of recurrence and treatment-related complications such as:
- Second malignancies
- Renal insufficiency
- Cardiotoxicity from anthracyclines
- Pulmonary fibrosis from radiation
Recommended Follow-Up Protocols
- Ultrasound every 3 months for the first 2 years
- Annual renal function tests
- Echocardiograms if doxorubicin was administered
- Genetic counseling for patients with syndromic or familial Wilms’ tumor
Recent Advances and Research in Wilms’ Tumor
Ongoing research is focused on refining risk stratification, reducing therapy-related toxicity, and targeting molecular pathways involved in tumorigenesis.
- Molecular profiling is increasingly used to identify actionable mutations
- WT1-targeted therapies are under exploration
- Minimally invasive nephrectomy techniques are improving surgical outcomes
- Clinical trials are investigating novel agents for relapsed or refractory disease
Wilms’ tumor represents one of the true successes in pediatric oncology, with survival rates continually improving due to advancements in surgery, chemotherapy, and radiation. Early diagnosis, accurate staging, and tailored therapy remain essential. With continued research, outcomes are expected to further improve, particularly in high-risk or relapsed disease.