Transthyretin familial amyloid polyneuropathy (TTR-FAP) is a rare, autosomal dominant, adult-onset neurodegenerative disease caused by mutations in the TTR (transthyretin) gene. The disease is characterized by progressive peripheral and autonomic neuropathy due to extracellular deposition of amyloid fibrils derived from misfolded TTR proteins.
TTR-FAP is a form of hereditary transthyretin amyloidosis (hATTR), typically involving the peripheral nervous system, cardiac tissue, and gastrointestinal tract. Without early intervention, the condition progresses rapidly, resulting in significant morbidity and early mortality.
Genetic Basis and Pathophysiology of TTR-FAP
Transthyretin is a tetrameric transport protein primarily synthesized in the liver. It binds and transports thyroxine (T4) and retinol-binding protein–vitamin A complex. Over 130 pathogenic mutations in the TTR gene on chromosome 18 have been identified, most notably the Val30Met (V30M) mutation.
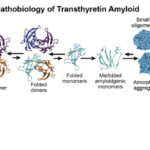
Pathogenic Cascade
- TTR Mutation
- Tetramer Destabilization
- Monomer Misfolding
- Amyloid Fibril Formation
- Deposition in Nerves and Organs
These deposits impair nerve conduction, damage autonomic neurons, and affect cardiomyocyte function, leading to polyneuropathy, cardiomyopathy, and autonomic dysfunction.
Epidemiology and Genetic Variants
TTR-FAP is endemic in certain regions:
- Portugal (V30M mutation)
- Japan
- Sweden
- Brazil
Onset typically occurs between ages 20 and 40 in endemic areas, but later-onset cases have been reported globally. Genetic anticipation and phenotypic variability are common, even among family members with the same mutation.
Clinical Manifestations of TTR-FAP
The clinical course is progressive, involving multiple systems. The initial symptoms are often subtle and easily misdiagnosed.
Neurological Symptoms
- Symmetric sensorimotor polyneuropathy: begins in the lower limbs
- Loss of pain and temperature sensation
- Paresthesias and distal muscle weakness
- Foot drop in advanced cases
Autonomic Dysfunction
- Orthostatic hypotension
- Urinary incontinence
- Erectile dysfunction
- Chronic diarrhea or constipation
- Gastroparesis
Cardiac Involvement
- Restrictive cardiomyopathy
- Conduction abnormalities
- Heart failure symptoms
Ocular and Renal Involvement
- Vitreous opacities
- Glaucoma
- Proteinuria in some variants
Diagnostic Criteria and Testing
Stepwise Diagnostic Approach
- Clinical Evaluation
- Family history of neuropathy or amyloidosis
- Neurological examination
- Nerve Conduction Studies
- Axonal polyneuropathy pattern
- Tissue Biopsy
- Congo red-positive amyloid deposits with apple-green birefringence under polarized light
- Genetic Testing
- Definitive confirmation via TTR gene sequencing
- Cardiac Assessment
- ECG and echocardiography
- Cardiac MRI and Technetium-99m scintigraphy for amyloid
- Autonomic Function Testing
- Heart rate variability
- Tilt-table testing
Differentiating TTR-FAP from Other Neuropathies
TTR-FAP must be distinguished from:
- Chronic inflammatory demyelinating polyneuropathy (CIDP)
- Diabetic neuropathy
- Idiopathic autonomic neuropathy
- AL amyloidosis
Key differentiators include family history, absence of inflammation in biopsies, and specific genetic mutations.
Staging and Prognosis
TTR-FAP is staged based on ambulatory capacity:
- Stage 1: Independent walking, mild symptoms
- Stage 2: Assistance required for walking
- Stage 3: Wheelchair-bound or bedridden
Without treatment, the average survival from symptom onset is 10 to 15 years, though cardiac forms may be more aggressive.
Treatment Options for TTR-FAP
Early intervention is critical. Therapeutic approaches aim to stabilize the TTR tetramer, reduce TTR production, or eliminate existing deposits.
1. TTR Stabilizers
- Tafamidis (Vyndaqel): Binds to TTR and prevents tetramer dissociation
- Diflunisal: NSAID with TTR-stabilizing properties
These agents delay neurological progression and are particularly effective in early-stage disease.
2. TTR Gene Silencers
- Patisiran (RNA interference)
- Inotersen (antisense oligonucleotide)
These agents reduce hepatic production of both wild-type and mutant TTR, leading to decreased amyloid burden.
3. Liver Transplantation
- Historically the only disease-modifying treatment
- Replaces the mutant TTR-producing liver
- Limited by cardiac and CNS amyloid accumulation, which persist post-transplant
4. Symptomatic Management
- Neuropathic pain: gabapentinoids, duloxetine
- Autonomic symptoms: midodrine, fludrocortisone
- Cardiac support: pacemaker insertion, heart failure management
Future Directions and Emerging Therapies
Research is ongoing to improve outcomes and slow disease progression:
- CRISPR-Cas9 gene editing: NTLA-2001 is in clinical trials
- Monoclonal antibodies targeting amyloid fibrils
- Second-generation gene silencers with improved safety profiles
These developments represent promising avenues for long-term disease control and potential cures.
Patient Support and Genetic Counseling
Given its hereditary nature, genetic counseling is essential for:
- Family screening
- Pre-symptomatic diagnosis
- Reproductive planning
Patient advocacy organizations offer education, support, and access to clinical trials.
Frequently Asked Questions:
What causes transthyretin familial amyloid polyneuropathy?
TTR-FAP is caused by mutations in the transthyretin gene that lead to misfolded proteins forming amyloid deposits.
How is TTR-FAP diagnosed?
Diagnosis involves genetic testing, tissue biopsy, nerve conduction studies, and systemic evaluation.
Can TTR-FAP be cured?
While there is no cure, therapies like Tafamidis, patisiran, and liver transplant can delay progression and improve quality of life.
Who is at risk for TTR-FAP?
Individuals with a family history of amyloidosis, especially in endemic regions, are at highest risk.
Is genetic testing available for family members?
Yes. TTR gene sequencing can identify carriers and help guide early intervention and counseling.
Transthyretin familial amyloid polyneuropathy represents a severe, progressive hereditary neuropathy with systemic involvement. Timely diagnosis, genetic screening, and evolving disease-modifying therapies have dramatically shifted the landscape of care. As precision medicine advances, targeted approaches hold promise to transform outcomes for patients with this once-fatal disease.