T-cell acute lymphoblastic leukemia (T-ALL) is a rare and aggressive subtype of acute lymphoblastic leukemia, primarily originating from immature T lymphocytes. Representing approximately 15% of pediatric and 25% of adult ALL cases, T-ALL progresses rapidly and demands immediate medical intervention. This hematologic malignancy often involves the bone marrow, blood, thymus, and, in advanced stages, other organs.
Pathophysiology and Cellular Origin
T-ALL originates from the malignant transformation of progenitor T-cells in the thymus. This transformation involves multiple genetic and epigenetic mutations, leading to uncontrolled proliferation and impaired differentiation.
Genetic Mutations in T-ALL
Key genetic alterations include:
- NOTCH1 mutations (in over 50% of cases)
- CDKN2A/2B deletions
- PTEN loss
- FBXW7 mutations
- IL7R mutations
These mutations disrupt pathways responsible for T-cell development and apoptosis, causing malignant T-cell proliferation.
Risk Factors and Epidemiology
T-ALL primarily affects children, adolescents, and young adults, with a male predominance. Risk factors include:
- Exposure to ionizing radiation
- Family history of hematologic malignancies
- Genetic syndromes (e.g., Li-Fraumeni syndrome, ataxia-telangiectasia)
While less common than B-cell ALL, T-ALL is known for its aggressive clinical course.
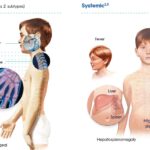
Clinical Presentation and Symptoms
Patients with T-ALL may present with a variety of symptoms, typically caused by bone marrow failure or mass effects from a mediastinal tumor.
Common Symptoms:
- Fatigue and pallor due to anemia
- Frequent infections from neutropenia
- Bleeding tendencies (petechiae, bruising)
- Enlarged thymus or mediastinal mass leading to respiratory distress
- Lymphadenopathy and hepatosplenomegaly
- Bone pain and joint discomfort
Infiltration into the central nervous system (CNS) may lead to neurological symptoms like headaches, seizures, or cranial nerve palsies.
Diagnosis of T-Cell Acute Lymphoblastic Leukemia
Timely diagnosis is critical. The diagnostic workup involves multiple assessments:
1. Complete Blood Count (CBC)
- Elevated white blood cell count (often >100,000/mm³)
- Anemia and thrombocytopenia
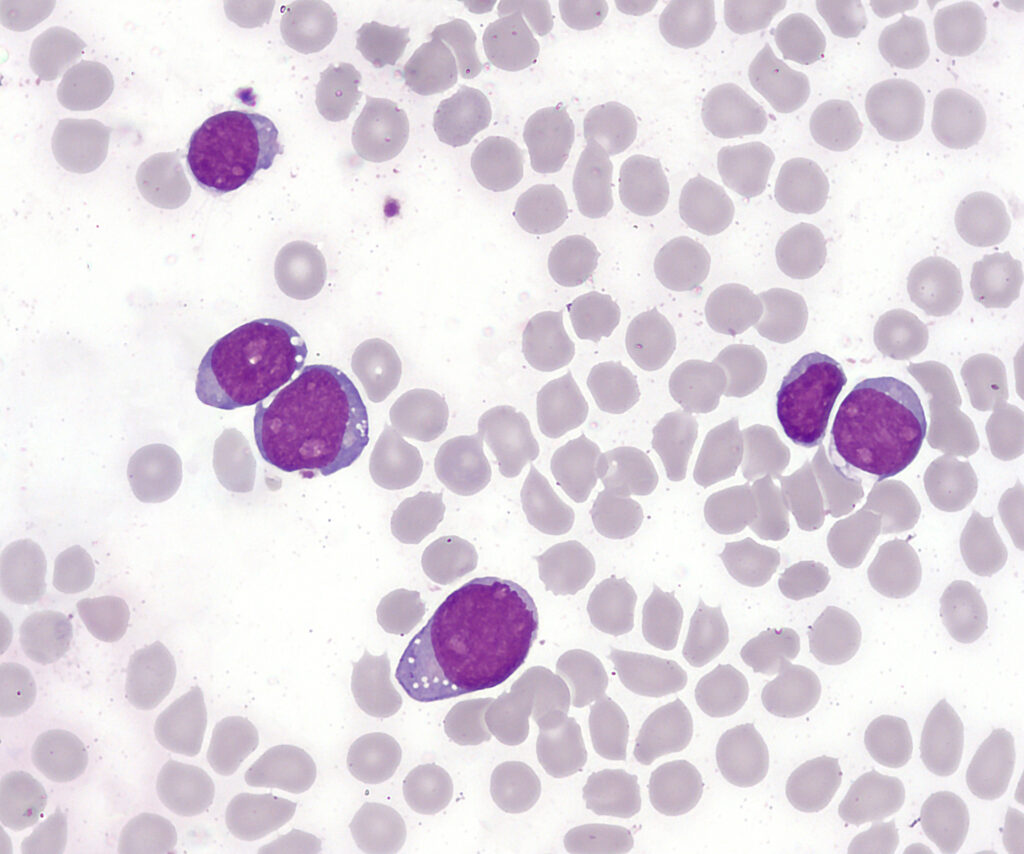
2. Peripheral Blood Smear
- Presence of lymphoblasts
3. Bone Marrow Biopsy
- Confirms >20% lymphoblasts in marrow
- Immunophenotyping via flow cytometry (CD3+, CD7+, CD2+, CD5+)
4. Cytogenetic and Molecular Analysis
- Detects chromosomal translocations and gene mutations
5. Imaging Studies
- Chest X-ray or CT for mediastinal mass
- MRI/CT of the brain for CNS involvement
6. Lumbar Puncture
- Evaluates CNS infiltration
Subtypes and Risk Stratification
T-ALL can be classified based on immunophenotypic features:
- Early T-precursor ALL (ETP-ALL): Poor prognosis, stem-cell–like profile
- Cortical T-ALL
- Mature T-ALL
Risk stratification is based on:
- Initial WBC count
- Age
- CNS involvement
- Early response to therapy (measured by minimal residual disease)
Treatment of T-ALL
Treatment of T-ALL involves intensive multi-agent chemotherapy and CNS prophylaxis. The treatment phases include:
1. Induction Therapy
Goal: Achieve complete remission (CR)
Drugs used: Vincristine, corticosteroids (dexamethasone or prednisone), anthracyclines (e.g., daunorubicin), L-asparaginase
2. Consolidation Therapy
Goal: Eliminate residual disease
Includes high-dose methotrexate, cytarabine, and 6-mercaptopurine
3. Maintenance Therapy
Goal: Prevent relapse
Duration: Up to 2 years
Regimen: Methotrexate and 6-mercaptopurine orally
4. CNS Prophylaxis
- Intrathecal chemotherapy (methotrexate ± cytarabine)
- Occasionally cranial irradiation for high-risk cases
5. Hematopoietic Stem Cell Transplantation (HSCT)
Considered in:
- Relapsed or refractory cases
- High-risk genetic features
- Persistent minimal residual disease
Emerging Therapies and Clinical Trials
Advancements in molecular genetics have driven new treatment approaches:
Targeted Therapies:
- Gamma-secretase inhibitors (targeting NOTCH1)
- JAK inhibitors
- PI3K/AKT/mTOR pathway inhibitors
Immunotherapies:
- CAR-T cells (limited success in T-ALL due to fratricide)
- Anti-CD38 and CD7 monoclonal antibodies
- Bispecific T-cell engagers (BiTEs)
Clinical trials continue to evaluate the safety and efficacy of these novel agents.
Prognosis and Survival Rates
Prognosis depends on age, genetic profile, and treatment response:
- Children: 5-year overall survival ~75–85%
- Adults: 5-year overall survival ~40–60%
- ETP-ALL and refractory disease carry worse outcomes
Follow-up and Long-term Monitoring
Survivors require ongoing surveillance for:
- Relapse (especially within the first 2–3 years)
- Treatment-related toxicities (cardiac, hepatic, neurocognitive)
- Secondary malignancies
- Psychosocial support and rehabilitation
Prevention and Genetic Counseling
While no direct preventive measures exist for T-ALL, genetic counseling is advised for families with hereditary cancer syndromes. Prompt evaluation of early symptoms remains the best tool for early detection and improved outcomes.
Prognostic Factors in T-ALL
| Prognostic Factor | Impact on Outcome |
|---|---|
| Age <1 or >10 years | Adverse |
| High initial WBC count | Adverse |
| CNS involvement at diagnosis | Adverse |
| Early response to therapy | Favorable |
| MRD negativity post-induction | Favorable |
| NOTCH1 mutation | Favorable (with chemotherapy) |
| ETP-ALL subtype | Adverse |
T-cell acute lymphoblastic leukemia remains a challenging malignancy marked by rapid progression and complex treatment protocols. However, with advancements in risk-adapted therapy and precision medicine, survival rates—especially in pediatric populations—have significantly improved. Ongoing research in genomics and immunotherapy holds promise for even more targeted and effective future treatments.