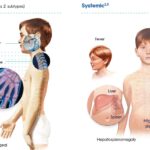
Systemic sclerosis (SSc), commonly known as scleroderma, is an autoimmune disease characterized by the hardening and tightening of the skin and connective tissues. However, one of the most significant and potentially life-threatening complications of systemic sclerosis is systemic sclerosis-associated interstitial lung disease (SS-ILD). This condition occurs when fibrosis, or scarring, develops in the lungs, leading to impaired pulmonary function.
SS-ILD is a major cause of morbidity and mortality in patients with systemic sclerosis, and its management is critical for improving patient outcomes. The disease can present with a variety of symptoms, ranging from mild respiratory issues to severe, life-limiting lung dysfunction. Early recognition and intervention are key to managing the disease effectively.
Understanding the Pathophysiology of Systemic Sclerosis-Associated Interstitial Lung Disease
Fibrosis and Lung Involvement in Systemic Sclerosis
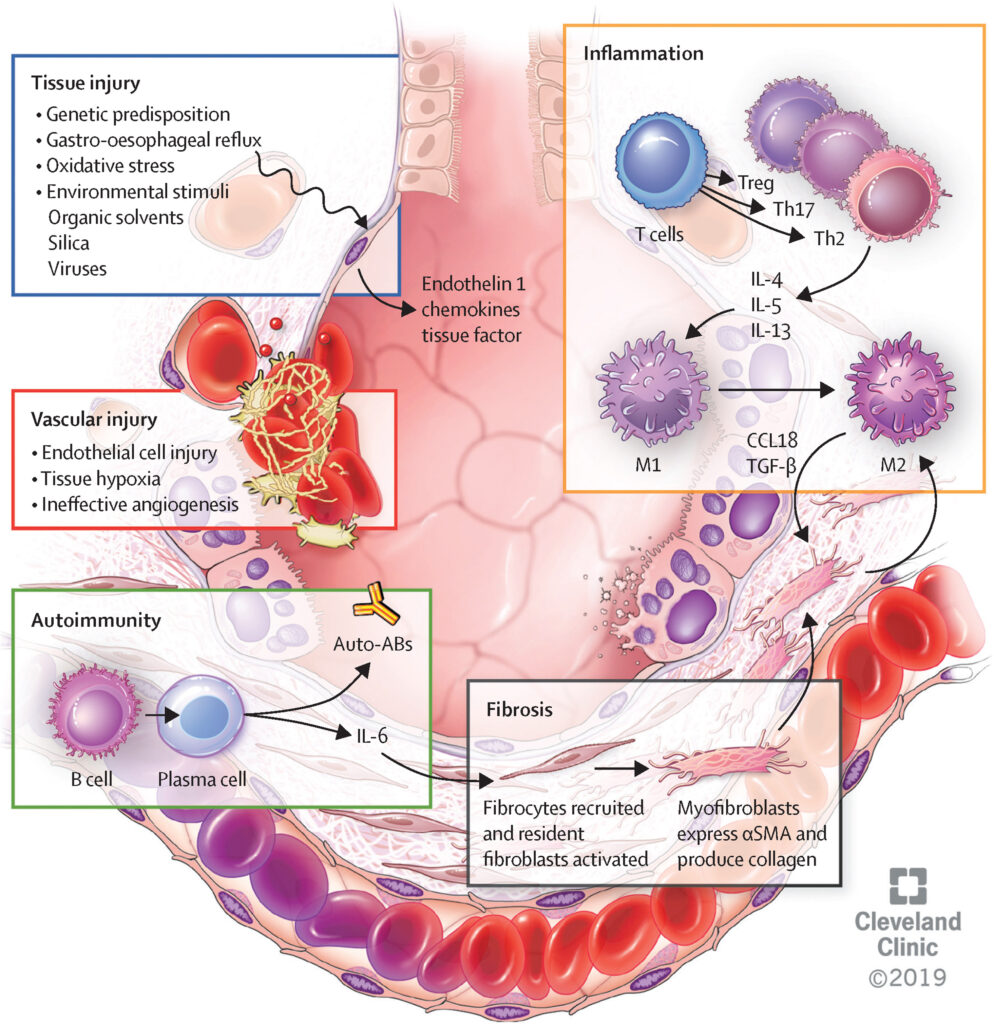
The pathophysiology of SS-ILD involves a complex interplay between immune dysregulation, vascular damage, and fibrosis. In systemic sclerosis, an overactive immune system triggers the production of collagen and other extracellular matrix proteins, which lead to tissue scarring. The lungs, being highly vascular and involved in gas exchange, are particularly susceptible to this fibrotic process.
The fibrotic changes in the lung parenchyma cause the interstitial tissue (the tissue surrounding the lung alveoli) to thicken, impairing the ability of oxygen to pass into the bloodstream. As the disease progresses, this scarring can lead to pulmonary fibrosis, a condition that significantly reduces lung function and can eventually cause respiratory failure.
Symptoms of Systemic Sclerosis-Associated Interstitial Lung Disease
Early Signs and Symptoms
The symptoms of SS-ILD can vary depending on the severity and progression of the disease. In the early stages, patients may experience mild and nonspecific respiratory symptoms that are often mistaken for other conditions. Some of the most common early symptoms include:
- Shortness of breath (dyspnea): This is one of the most common complaints and may occur during physical activity or even at rest in severe cases.
- Chronic dry cough: A persistent dry cough without an obvious cause, such as an infection, can be an early indicator of lung involvement.
- Fatigue: As lung function declines, patients may feel more fatigued due to the reduced oxygen levels in their blood.
Advanced Symptoms
As the disease progresses, the symptoms of SS-ILD can worsen and lead to more severe respiratory issues. These include:
- Severe shortness of breath: Patients may experience difficulty breathing even while at rest.
- Hypoxemia: Low oxygen levels in the blood, often resulting in confusion, dizziness, and cyanosis (a bluish tint to the skin, especially around the lips and fingertips).
- Pulmonary hypertension: As the lungs become more damaged, the blood pressure within the pulmonary arteries may rise, leading to additional cardiovascular complications.
- Worsening cough and sputum production: The development of mucus production or changes in the nature of the cough may signal worsening of lung involvement.
Diagnosis of Systemic Sclerosis-Associated Interstitial Lung Disease
Clinical Evaluation and History
Diagnosing SS-ILD begins with a detailed clinical evaluation. A patient’s history of systemic sclerosis, along with any symptoms of lung involvement, should raise suspicion. Patients with systemic sclerosis who present with respiratory symptoms should be carefully monitored for signs of interstitial lung disease.
Diagnostic Tests
Several diagnostic tests are used to confirm the presence of SS-ILD and assess the extent of lung involvement:
- Pulmonary Function Tests (PFTs): These tests measure lung capacity and the efficiency of gas exchange. A restrictive pattern of lung disease, where lung volume is reduced but airflow remains normal, is often seen in SS-ILD.
- High-Resolution Computed Tomography (HRCT): HRCT scans are considered the gold standard for visualizing lung changes associated with interstitial lung disease. They can detect early-stage fibrosis, ground-glass opacities, and honeycombing in the lungs, which are characteristic of SS-ILD.
- Arterial Blood Gas (ABG) Analysis: ABG tests measure oxygen levels in the blood. Hypoxemia, or low oxygen levels, is a common finding in patients with advanced SS-ILD.
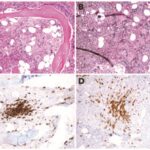
- Lung Biopsy: In some cases, a lung biopsy may be necessary to obtain tissue samples and definitively diagnose the extent of fibrosis or inflammation. However, this is usually reserved for patients where the diagnosis is unclear after non-invasive testing.
- Serological Testing: Blood tests to identify specific autoimmune markers, such as anti-topoisomerase I antibodies or anti-centromere antibodies, can help confirm the diagnosis of systemic sclerosis and distinguish it from other forms of lung disease.
Treatment Options for Systemic Sclerosis-Associated Interstitial Lung Disease
Medications and Therapies
- Immunosuppressive Therapy: Medications that suppress the overactive immune system are commonly used in the treatment of SS-ILD. Cyclophosphamide, mycophenolate mofetil, and azathioprine are frequently prescribed to reduce inflammation and fibrosis in the lungs.
- Antifibrotic Drugs: The recent introduction of antifibrotic medications such as nintedanib and pirfenidone has shown promise in slowing the progression of pulmonary fibrosis. These drugs are used to target the fibrotic processes and help preserve lung function.
- Corticosteroids: In some cases, corticosteroids may be used to manage inflammation in the early stages of SS-ILD, but their use is limited due to potential long-term side effects, especially in the setting of systemic sclerosis.
- Oxygen Therapy: As the disease progresses and pulmonary function deteriorates, oxygen therapy may be necessary to maintain adequate oxygen levels in the blood and prevent complications related to hypoxemia.
- Pulmonary Rehabilitation: A comprehensive program involving exercise, education, and breathing techniques can help patients improve their quality of life and manage symptoms of shortness of breath.
Lung Transplantation
In severe cases where pulmonary function is severely compromised and other treatments are no longer effective, lung transplantation may be considered. This is typically reserved for patients with end-stage SS-ILD who have failed other therapeutic options.
Prognosis and Life Expectancy
The prognosis of systemic sclerosis-associated interstitial lung disease depends on several factors, including the severity of lung involvement, the effectiveness of treatment, and the progression of the underlying systemic sclerosis. Early diagnosis and aggressive management can help slow the progression of the disease and improve outcomes. However, without treatment, SS-ILD can lead to significant respiratory compromise and a reduced quality of life.
Patients with mild SS-ILD may experience a stable course for years, while those with more severe fibrosis or associated complications such as pulmonary hypertension may have a worse outlook.
Survival Rates
Survival rates for SS-ILD vary widely depending on disease severity and response to treatment. Studies have shown that patients with moderate to severe fibrosis may have a reduced life expectancy, particularly if pulmonary hypertension or respiratory failure occurs.
Managing Systemic Sclerosis-Associated Interstitial Lung Disease
Regular Monitoring and Follow-Up
Due to the progressive nature of SS-ILD, patients require regular follow-up with pulmonologists to monitor lung function and adjust treatment plans. Pulmonary function tests, HRCT scans, and oxygen saturation measurements are key to tracking disease progression and the effectiveness of ongoing treatments.
Lifestyle Modifications
Patients with SS-ILD should adopt a healthy lifestyle to support overall lung health. This includes:
- Avoiding smoking and exposure to respiratory irritants.
- Maintaining a healthy weight to reduce strain on the lungs.
- Engaging in mild physical activity as tolerated to strengthen respiratory muscles.
Systemic sclerosis-associated interstitial lung disease is a serious complication of systemic sclerosis that can significantly impact a patient’s quality of life. Early diagnosis, prompt treatment, and ongoing monitoring are essential to managing the disease effectively. With advancements in therapies aimed at reducing fibrosis and improving lung function, patients now have better options for managing SS-ILD and achieving a more favorable long-term outcome.