Systemic anaplastic large cell lymphoma (sALCL) is a rare but aggressive subtype of non-Hodgkin lymphoma (NHL), classified under mature peripheral T-cell lymphomas. It is characterized by large, pleomorphic lymphoid cells expressing CD30—a critical diagnostic and therapeutic marker. sALCL comprises two major clinical and molecular subtypes based on the presence or absence of an anaplastic lymphoma kinase (ALK) gene rearrangement: ALK-positive sALCL and ALK-negative sALCL.
ALK-Positive sALCL
- Genetic hallmark: Translocation t(2;5)(p23;q35) resulting in NPM1-ALK fusion gene
- Demographics: Primarily affects children and young adults
- Prognosis: Generally favorable with high response to chemotherapy
- Immunophenotype: CD30+, ALK+, CD3−/+
ALK-Negative sALCL
- Absence of ALK gene rearrangement
- Demographics: More common in older adults
- Prognosis: Less favorable, with increased relapse risk
- Molecular heterogeneity: Includes DUSP22 and TP63 rearrangements—significant prognostic indicators
Pathophysiology and Molecular Features
Systemic ALCL originates from activated mature T-cells, often of cytotoxic phenotype. ALK-positive sALCL features constitutive tyrosine kinase activity due to ALK fusion proteins, leading to uncontrolled cellular proliferation. Conversely, ALK-negative sALCL lacks this mutation but may harbor genetic alterations such as:
- DUSP22 rearrangements (better outcomes)
- TP63 rearrangements (poor outcomes)
- JAK/STAT pathway mutations
- Epigenetic deregulations
These molecular insights are increasingly guiding targeted therapies and risk stratification.
Clinical Presentation of Systemic ALCL
sALCL typically presents with generalized symptoms and widespread lymphadenopathy. B-symptoms and extranodal involvement are common.
Common Signs and Symptoms:
- Painless lymph node enlargement (especially cervical, axillary)
- Fever, night sweats, unexplained weight loss
- Hepatosplenomegaly
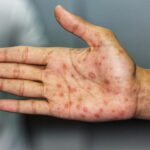
- Cutaneous involvement in some cases
- Bone marrow infiltration (rare but possible)
- In pediatric ALK+ cases: mediastinal mass or systemic symptoms may dominate
Diagnostic Criteria for sALCL
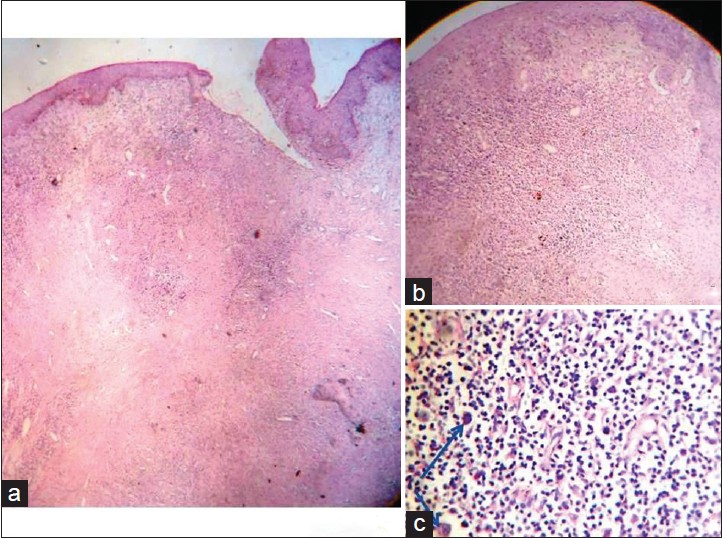
Histopathological Features:
- Large pleomorphic cells with horseshoe- or kidney-shaped nuclei (hallmark cells)
- Abundant cytoplasm, frequent mitotic figures
- Strong membranous and Golgi staining for CD30
Immunophenotyping:
- Positive: CD30, EMA, cytotoxic granule proteins (TIA-1, granzyme B), ALK (in ALK+)
- Variable: CD3, CD4, CD43
- Negative: CD15 (helps distinguish from Hodgkin lymphoma)
Additional Diagnostics:
- ALK immunohistochemistry: Distinguishes ALK+ from ALK−
- FISH/PCR for ALK, DUSP22, TP63 rearrangements
- PET-CT and bone marrow biopsy: For staging
Staging System: Ann Arbor Classification
sALCL follows the Ann Arbor staging system used for lymphomas:
| Stage | Description |
|---|---|
| I | Single lymph node region or single extralymphatic site |
| II | Two or more lymph node regions on the same side of the diaphragm |
| III | Lymph nodes on both sides of the diaphragm |
| IV | Disseminated involvement of one or more extralymphatic organs |
Treatment Strategies for Systemic ALCL
First-Line Treatment
- CHOP chemotherapy: Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone
- Brentuximab vedotin (BV) + CHOP (BV-CHP): Emerging standard, particularly in CD30+ cases
ALK-Positive sALCL
- Highly responsive to CHOP or BV-CHP
- Long-term survival >70%
- Pediatric regimens may include APO (doxorubicin, prednisone, vincristine, methotrexate)
ALK-Negative sALCL
- Often requires intensified regimens
- Clinical trials with novel agents or autologous stem cell transplant (ASCT) in first remission
Relapsed/Refractory Disease
- Brentuximab vedotin: Anti-CD30 antibody-drug conjugate
- ALK inhibitors (e.g., crizotinib) for ALK+ relapses
- Stem cell transplantation: Auto or allo-HSCT in eligible patients
- Novel agents: Immune checkpoint inhibitors, JAK inhibitors, epigenetic therapies under investigation
Prognosis and Survival Rates
ALK-Positive sALCL
- 5-year overall survival (OS): 70–90%
- Favorable prognostic indicators: young age, good performance status, early-stage disease
ALK-Negative sALCL
- 5-year OS: ~49–60%
- TP63 rearrangement: very poor outcomes
- DUSP22 rearrangement: outcomes comparable to ALK+ cases
Prognostic Factors:
- IPI score (age, stage, LDH, performance status, extranodal disease)
- Molecular subtype (ALK, DUSP22, TP63)
- Response to frontline therapy
Pediatric Considerations
Pediatric systemic ALCL (primarily ALK+) presents with more widespread disease but has superior outcomes compared to adults. Multimodal therapy including CHOP-based regimens and BV has significantly improved pediatric survival rates.
- Event-free survival: ~75–80%
- Close monitoring for long-term toxicities and relapse is essential
Future Directions in Research and Therapy
- Targeted therapies: Expanding role of ALK inhibitors and anti-CD30 agents
- Immunotherapy: Checkpoint blockade (PD-1/PD-L1), CAR-T cell therapy
- Biomarker development: For better prognostication and personalized treatment
- Epigenetic modifiers: HDAC and EZH2 inhibitors in clinical trials
Frequently Asked Questions:
Q1. Is systemic ALCL curable?
Yes, especially in ALK-positive cases. Early detection and appropriate therapy significantly improve outcomes.
Q2. What is the difference between systemic and cutaneous ALCL?
Systemic ALCL involves nodal and systemic sites, while cutaneous ALCL is localized to the skin and usually has a better prognosis.
Q3. How is ALK status determined?
Immunohistochemistry and FISH are standard methods to assess ALK protein expression or gene rearrangement.
Q4. Are there any targeted therapies for systemic ALCL?
Yes. Brentuximab vedotin and ALK inhibitors are approved for specific subtypes and disease stages.
Q5. What are the common side effects of treatment?
Chemotherapy and targeted therapy may cause fatigue, neuropathy, neutropenia, and infection risks, requiring close monitoring.