Synovial sarcoma is a rare and aggressive soft tissue malignancy that arises from mesenchymal cells. Despite its name, it does not originate from synovial tissue but is most commonly located near joints in the extremities, particularly in the lower limbs. It accounts for approximately 5–10% of all soft tissue sarcomas and frequently affects adolescents and young adults aged 15 to 40.
This malignancy is characterized by a distinct chromosomal translocation t(X;18)(p11.2;q11.2), which results in the SS18-SSX fusion gene—a diagnostic hallmark and therapeutic target.
Clinical Presentation and Symptoms
Synovial sarcoma typically presents as a slow-growing, deep-seated mass adjacent to a joint, often mistaken for a benign condition. Clinical symptoms include:
- Localized swelling or lump near a joint
- Pain or tenderness if the tumor compresses nearby nerves
- Reduced range of motion
- Asymptomatic masses detected incidentally
Late-stage presentations may involve systemic symptoms such as fatigue, weight loss, or metastasis-related complications, particularly in the lungs.
Histological and Molecular Subtypes
Synovial sarcoma exhibits three main histological variants:
- Biphasic Type: Composed of both epithelial and spindle cell components.
- Monophasic Type: Primarily spindle cells; more common.
- Poorly Differentiated Type: Aggressive with high mitotic activity.
All types share the SS18-SSX fusion gene, detected via molecular diagnostics such as RT-PCR or FISH.
Synovial Sarcoma Pathogenesis: SS18-SSX Fusion Mechanism
The oncogenic fusion occurs when the SS18 gene on chromosome 18 fuses with SSX1, SSX2, or SSX4 on the X chromosome. This chimeric protein disrupts chromatin remodeling and drives aberrant transcription.
This molecular event is pathognomonic and guides diagnostic and therapeutic strategies.
Diagnostic Approach
Imaging Studies
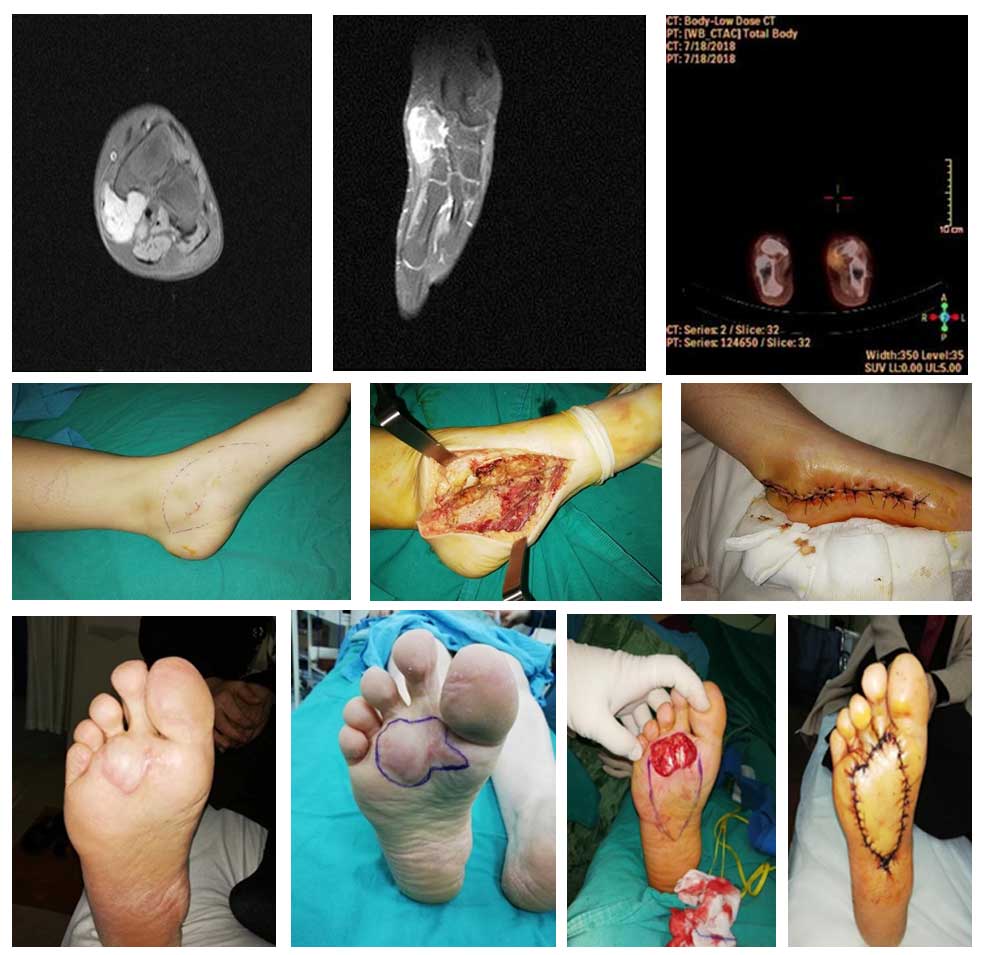
- MRI is the gold standard for local tumor characterization, revealing well-defined masses with heterogeneous enhancement.
- CT Scans are preferred for assessing pulmonary metastasis.
- PET-CT may aid in detecting occult metastases and evaluating treatment response.
Biopsy and Histopathology
- Core needle biopsy under imaging guidance is preferred.
- Immunohistochemical markers include TLE1, BCL-2, and CD99, though definitive diagnosis requires molecular confirmation of the SS18-SSX translocation.
Synovial Sarcoma Staging and Prognostic Factors
Staging follows the AJCC TNM system, incorporating tumor size, nodal involvement, and metastasis. Prognosis is influenced by:
- Tumor size >5 cm (worse outcome)
- High histological grade
- Presence of metastases at diagnosis
- Fusion type (SS18-SSX1 linked to poorer prognosis than SSX2)
Treatment Options for Synovial Sarcoma
Surgical Resection
Wide local excision with negative margins is the cornerstone of treatment. Limb-sparing surgery is preferred over amputation whenever feasible.
Radiation Therapy
Adjuvant radiotherapy is used to reduce local recurrence, especially in high-grade or margin-positive tumors.
Chemotherapy
Chemotherapeutic regimens typically involve:
- Ifosfamide and Doxorubicin as frontline agents
- Response rates vary, but systemic therapy is critical in metastatic or high-risk cases
Targeted and Experimental Therapies
- Tazemetostat (EZH2 inhibitor) and pazopanib (multi-tyrosine kinase inhibitor) are under investigation.
- Clinical trials targeting SS18-SSX fusion mechanisms and immune checkpoint inhibitors offer promise.
Prognosis and Survival Rates
The 5-year overall survival rate varies based on stage and response to treatment:
| Stage | 5-Year Survival Rate |
|---|---|
| Localized | 70–80% |
| Regional (nodal involvement) | 50–60% |
| Metastatic | 20–30% |
Early detection, complete surgical excision, and multimodal therapy significantly improve outcomes.
Pediatric and Adolescent Considerations
Synovial sarcoma is among the most common non-rhabdomyosarcoma soft tissue sarcomas in children and adolescents. Treatment protocols often follow pediatric-specific cooperative group guidelines, balancing oncologic control with long-term functional outcomes.
Follow-Up and Long-Term Monitoring
Patients require structured surveillance due to high risk of late recurrences and metastases:
- Every 3–6 months for the first 2 years, including chest imaging
- Annually after 5 years, especially in high-risk cases
Functional rehabilitation and psychosocial support play a key role in long-term recovery.
Future Directions in Synovial Sarcoma Research
- Liquid biopsies for early recurrence detection using circulating tumor DNA (ctDNA)
- Fusion-specific vaccines and T-cell therapies under investigation
- Next-generation sequencing to uncover secondary mutations and resistance mechanisms
Frequently Asked Questions:
Q1. What causes synovial sarcoma?
It is caused by a chromosomal translocation resulting in the SS18-SSX fusion gene, leading to abnormal cell proliferation.
Q2. Is synovial sarcoma curable?
Yes, when detected early and treated aggressively with surgery and adjunct therapies, long-term remission is possible.
Q3. How aggressive is synovial sarcoma?
It is considered an intermediate to high-grade malignancy with a high risk of recurrence and metastasis if not treated appropriately.
Q4. What is the role of chemotherapy in synovial sarcoma?
Chemotherapy is used in high-grade, large, or metastatic tumors and can improve survival and reduce recurrence risk.
Q5. Can synovial sarcoma metastasize?
Yes, the most common site of metastasis is the lungs, followed by lymph nodes and bone.