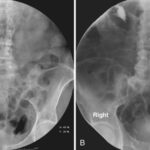
Subependymal giant cell astrocytomas (SEGAs) are rare, slow-growing brain tumors typically associated with tuberous sclerosis complex (TSC). TSC is a genetic disorder characterized by the growth of benign tumors in various organs, most notably in the brain, kidneys, heart, and skin. SEGAs represent one of the most common brain tumor types found in patients with TSC, and their presence significantly impacts clinical management and patient outcomes.
This article provides an in-depth exploration of subependymal giant cell astrocytomas, detailing their relationship with tuberous sclerosis, clinical manifestations, diagnostic methods, treatment options, and management strategies.
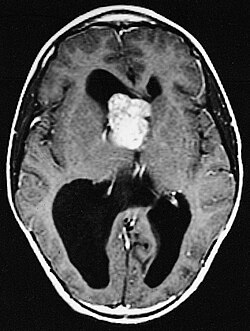
Understanding Subependymal Giant Cell Astrocytomas (SEGA)
Subependymal giant cell astrocytomas are a form of glioma, a tumor that arises from glial cells in the brain. These tumors are typically located near the ventricular system, particularly the lateral ventricles, and often present in childhood or early adulthood. SEGAs are characterized by their slow growth and low-grade malignancy, making them distinct from other more aggressive brain tumors.
In TSC, SEGAs are caused by mutations in either the TSC1 or TSC2 genes, which regulate cell growth. These mutations lead to abnormal cell proliferation in the brain, resulting in the development of SEGAs. Although SEGAs are benign, their location and growth can cause significant neurological impairment due to increased intracranial pressure, obstruction of cerebrospinal fluid flow, and other complications.
Key Features of SEGA:
- Location: Typically near the lateral ventricles or other subependymal regions of the brain.
- Size: They can range from small to large, sometimes causing hydrocephalus due to their proximity to the ventricular system.
- Growth Rate: SEGAs grow slowly but can lead to substantial neurological symptoms if left untreated.
Subependymal Giant Cell Astrocytomas and Tuberous Sclerosis Complex (TSC)
Tuberous sclerosis complex is an autosomal dominant genetic disorder caused by mutations in the TSC1 or TSC2 genes. These genes encode proteins (hamartin and tuberin) that act as tumor suppressors. When these genes are mutated, the control over cell growth and division is lost, leading to the formation of benign tumors, or hamartomas, in various tissues.
One of the most notable manifestations of TSC is the development of SEGAs in the brain. It is estimated that around 15-20% of individuals with TSC will develop subependymal giant cell astrocytomas. These tumors are strongly associated with the TSC1 and TSC2 mutations and serve as a significant diagnostic marker for the condition.
TSC and SEGAs: Pathophysiological Link
The growth of SEGAs in TSC is directly linked to dysregulation of the mTOR (mechanistic target of rapamycin) signaling pathway, which plays a critical role in regulating cell growth and metabolism. Mutations in the TSC1 or TSC2 genes result in abnormal mTOR activation, leading to the uncontrolled cell proliferation characteristic of SEGAs.
Clinical Presentation of Subependymal Giant Cell Astrocytomas
The clinical presentation of subependymal giant cell astrocytomas can vary depending on the tumor’s size, location, and rate of growth. Symptoms are primarily related to increased intracranial pressure (ICP) or the direct effects of the tumor on adjacent brain structures. Common presenting symptoms include:
1. Seizures
- Seizures are the most frequent symptom in patients with SEGAs, occurring in approximately 60-80% of cases. These seizures are often focal, reflecting the tumor’s location within the brain.
2. Headaches
- Headaches resulting from elevated intracranial pressure are common, particularly if the tumor is large or obstructs cerebrospinal fluid flow.
3. Hydrocephalus
- Larger SEGAs may obstruct the flow of cerebrospinal fluid, leading to hydrocephalus, a condition characterized by an abnormal accumulation of fluid in the ventricles of the brain.
4. Cognitive or Motor Impairment
- Depending on the tumor’s location, patients may experience cognitive decline, motor deficits, or visual disturbances.
5. Developmental Delays in Children
- Children with TSC and SEGAs may experience developmental delays, particularly in areas of motor and cognitive function.
Diagnosing Subependymal Giant Cell Astrocytomas
Accurate diagnosis of SEGAs is crucial for effective management and treatment. Several diagnostic methods are employed to identify the presence and extent of the tumor.
1. Magnetic Resonance Imaging (MRI)
MRI is the gold standard for diagnosing subependymal giant cell astrocytomas. SEGAs appear as well-defined, enhancing masses near the ventricles, often with areas of necrosis or cystic changes. Contrast-enhanced MRI can provide detailed imaging of the tumor’s location and help assess its relationship with surrounding structures.
2. Computed Tomography (CT) Scanning
Although MRI is preferred, CT scans may be used in emergencies or if MRI is unavailable. SEGAs may show up as hyperdense lesions in the brain, but CT scans are less sensitive than MRI for detecting these tumors, especially in the early stages.
3. Genetic Testing
Genetic testing to identify mutations in the TSC1 or TSC2 genes is an essential diagnostic tool for confirming tuberous sclerosis and its association with SEGAs. Genetic testing is especially useful for early diagnosis in patients without clear clinical symptoms.
4. Electroencephalogram (EEG)
EEG may be performed to assess seizure activity in patients with SEGAs, especially those presenting with focal seizures.
Treatment Options for Subependymal Giant Cell Astrocytomas
The management of SEGAs depends on the tumor’s size, symptoms, and potential for progression. Treatment may include surgical intervention, medical therapy, or a combination of both.
1. Surgical Resection
Surgical removal of the tumor is often the treatment of choice for patients with symptomatic SEGAs, particularly those causing hydrocephalus or intractable seizures. Surgery can significantly alleviate symptoms and prevent further neurological deterioration. However, complete resection may not always be possible due to the tumor’s location near critical brain structures.
2. Medical Therapy: mTOR Inhibitors
Recent advances in medical therapy have led to the use of mTOR inhibitors, such as everolimus, in the treatment of SEGAs. Everolimus has shown promising results in reducing tumor size and improving neurological function in patients with TSC-associated SEGAs. mTOR inhibitors work by targeting the dysregulated mTOR signaling pathway that drives tumor growth in TSC.
3. Shunt Placement for Hydrocephalus
In cases where SEGAs cause hydrocephalus, surgical placement of a shunt may be necessary to drain excess cerebrospinal fluid and relieve pressure on the brain.
Prognosis and Long-Term Management
The prognosis for patients with subependymal giant cell astrocytomas varies depending on the tumor’s size, location, and response to treatment. Early diagnosis and intervention are critical for improving long-term outcomes. With appropriate treatment, many patients experience significant improvement in neurological function and quality of life.
Follow-Up Care
Ongoing monitoring of patients with SEGAs is essential to detect tumor recurrence or progression. Regular MRI scans and clinical assessments are necessary to evaluate the effectiveness of treatment and manage any complications.
Subependymal giant cell astrocytomas associated with tuberous sclerosis represent a significant clinical challenge in neuro-oncology. Although these tumors are benign, their potential for causing severe neurological impairment necessitates early diagnosis and effective treatment. Advances in genetic testing, imaging technologies, and targeted therapies have improved outcomes for patients with SEGAs, offering hope for better management and long-term prognosis.