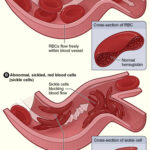
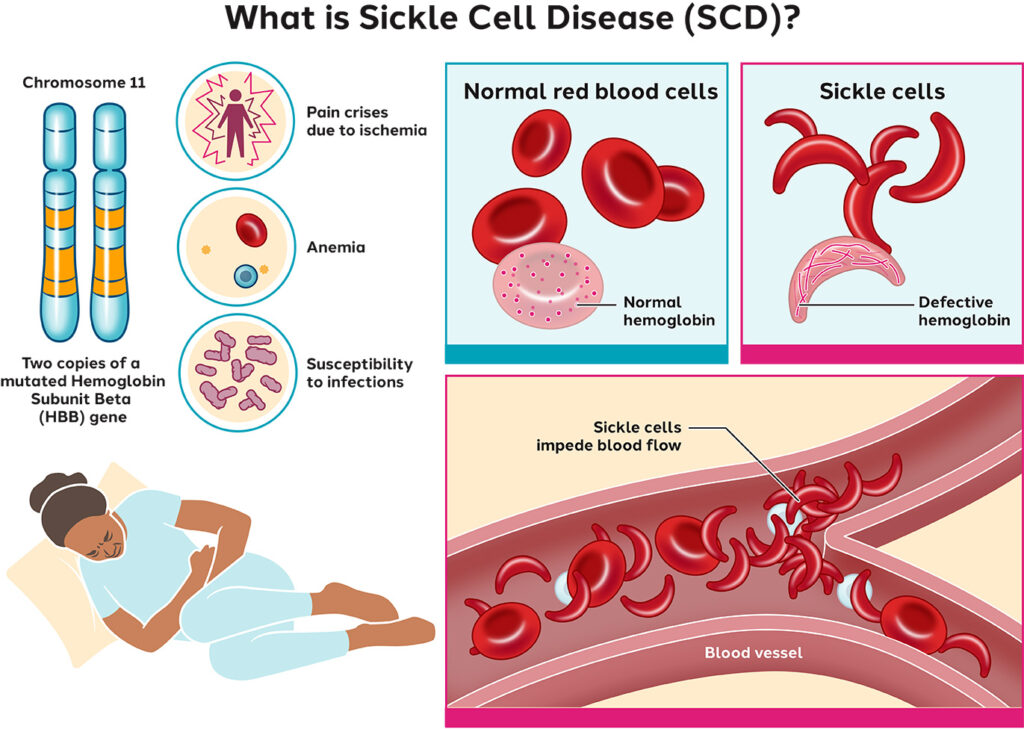
Sickle cell disease (SCD) is a group of inherited red blood cell disorders caused by a mutation in the HBB gene leading to the production of abnormal hemoglobin known as hemoglobin S. This mutation causes red blood cells to assume a rigid, sickle-like shape, impairing their ability to transport oxygen efficiently and leading to recurrent episodes of vascular blockage and systemic complications.
Genetics and Inheritance of Sickle Cell Disease
SCD follows an autosomal recessive inheritance pattern. A child must inherit one defective gene from each parent to develop the disease. Individuals with only one mutated gene are carriers (sickle cell trait) and typically do not display symptoms but can pass the gene to offspring.
Pathophysiology of Sickle Cell Disease
In SCD, hemoglobin S polymerizes under deoxygenated conditions, distorting red blood cells into sickle shapes. These malformed cells are fragile, leading to hemolysis, and are prone to adhering to vascular endothelium, causing vaso-occlusion.
Key Pathological Features:
- Hemolytic anemia due to short RBC lifespan (~20 days)
- Vascular inflammation and ischemia
- Organ damage from repeated oxygen deprivation
- Increased susceptibility to infection
Clinical Manifestations of Sickle Cell Disease
Common Symptoms
- Chronic anemia
- Fatigue
- Shortness of breath
- Pallor
- Jaundice
Sickle Cell Crises
- Vaso-occlusive crisis (pain crisis): Sudden, severe pain due to microvascular obstruction.
- Acute chest syndrome: Chest pain, fever, respiratory distress, and pulmonary infiltrates.
- Splenic sequestration: Rapid splenic enlargement and severe anemia.
- Aplastic crisis: Temporary cessation of RBC production, often triggered by parvovirus B19.
Chronic Complications
- Stroke (especially in children)
- Avascular necrosis (e.g., femoral head)
- Chronic kidney disease
- Retinopathy and vision loss
- Leg ulcers
- Delayed growth and puberty
Diagnosis of Sickle Cell Disease
Early detection is critical, especially in newborns, to prevent complications. Diagnostic tools include:
Screening and Confirmatory Tests
- Newborn screening (heel prick test)
- Hemoglobin electrophoresis – identifies the presence of HbS
- High-performance liquid chromatography (HPLC)
- Genetic testing – detects HBB mutations
Supportive Laboratory Findings
- Elevated reticulocyte count
- Elevated bilirubin and LDH
- Low hemoglobin and hematocrit
- Peripheral smear showing sickled cells and Howell-Jolly bodies
Treatment and Management of Sickle Cell Disease
1. Pharmacologic Therapies
| Treatment | Mechanism | Purpose |
|---|---|---|
| Hydroxyurea | Increases fetal hemoglobin (HbF) | Reduces frequency of crises |
| Voxelotor | Inhibits HbS polymerization | Improves hemoglobin levels |
| Crizanlizumab | P-selectin inhibitor | Reduces vaso-occlusive events |
| L-glutamine | Reduces oxidative stress | Lowers pain crisis occurrence |
| Penicillin prophylaxis | Prevents pneumococcal infections | Essential in young children |
2. Blood Transfusions
Used to treat severe anemia, stroke prevention, and acute complications. Chronic transfusion may lead to iron overload, necessitating chelation therapy.
3. Curative Treatment: Bone Marrow or Stem Cell Transplant
The only known cure. Most effective when performed in children with a matched sibling donor. Limitations include donor availability and transplant-related risks.
Supportive and Preventive Care
Routine Preventive Measures
- Routine vaccinations (pneumococcal, meningococcal, influenza)
- Annual transcranial Doppler ultrasound (in children to assess stroke risk)
- Adequate hydration and pain management protocols
- Regular monitoring for organ function (renal, hepatic, pulmonary)
Lifestyle and Psychosocial Support
- Nutritional counseling to address growth delays
- Mental health support for chronic pain and depression
- School and vocational support services
Emerging Therapies and Research in SCD
Innovative treatments targeting the genetic root or downstream effects of SCD are in development.
Gene Therapy
- CRISPR-based gene editing to reactivate fetal hemoglobin
- Lentiviral vectors for gene replacement
- Ongoing clinical trials show promising results
Novel Pharmacologic Agents
- Anti-adhesion molecules
- Antioxidants
- Hemoglobin modulators
Sickle Cell Disease in Global Context
Epidemiology
- Affects approximately 20 million people globally
- Highest prevalence in sub-Saharan Africa, India, and Middle Eastern countries
- Carrier rates in African-Americans ~8–10%
Public Health Considerations
- Neonatal screening programs are critical for early diagnosis
- Education and access to care reduce mortality rates
- Global disparities in treatment access remain a concern
Living with Sickle Cell Disease: Long-Term Outlook
With comprehensive care, many individuals with SCD live into adulthood. Advances in treatment have significantly improved life expectancy and quality of life. Multidisciplinary management, access to hematology specialists, and education play key roles in long-term outcomes.
Sickle cell disease is a life-altering inherited condition marked by complex pathophysiology and systemic complications. Early diagnosis, preventive care, and emerging therapies offer significant hope. Through ongoing research and global health initiatives, the burden of SCD can be mitigated, improving survival and life quality for millions affected worldwide.