Cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD) is a rare X-linked neurodevelopmental condition marked by early-onset, refractory seizures and profound developmental delays. The hallmark feature of CDD is epilepsy, typically manifesting within the first few months of life. These seizures are often resistant to standard anti-seizure medications and contribute significantly to the disorder’s morbidity.
Genetic and Molecular Basis of CDKL5-Related Seizures
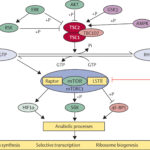
CDKL5 Gene Function and Pathogenic Mutations
The CDKL5 gene, located on the X chromosome (Xp22), encodes a serine/threonine kinase crucial for postnatal brain development. It plays a regulatory role in:
- Neuronal maturation
- Synaptic plasticity
- Dendritic arborization
Mutations leading to CDKL5 loss-of-function disrupt these processes, producing widespread cortical hyperexcitability and facilitating seizure generation.
Inheritance and Gender Impact
Due to X-linked dominance, CDKL5 mutations predominantly affect females. Males with CDKL5 mutations often exhibit more severe phenotypes, including intractable epilepsy and higher mortality rates.
Clinical Presentation of Seizures in CDKL5 Deficiency Disorder
Early-Onset Epileptic Seizures
The majority of patients develop seizures within the first 3 months of life. Common seizure types include:
- Infantile spasms (West syndrome)
- Tonic seizures
- Focal seizures
- Myoclonic seizures
- Epileptic spasms transitioning to Lennox-Gastaut-like patterns
These seizures typically occur in clusters, may be misdiagnosed as benign neonatal events initially, and tend to worsen over time.
EEG Characteristics
Electroencephalography findings often reveal:
- Multifocal epileptiform discharges
- Hypsarrhythmia in infantile spasms
- Diffuse background slowing
- Evolving patterns toward Lennox-Gastaut syndrome features
Diagnostic Criteria and Genetic Testing
Recognizing CDKL5 Syndrome in Epileptic Encephalopathies
CDD should be suspected in infants presenting with:
- Seizures beginning before 6 months of age
- Developmental regression or stagnation
- Hypotonia, cortical visual impairment, and movement disorders
Genetic Confirmation
- Whole-exome sequencing (WES) or targeted CDKL5 gene panel confirms diagnosis
- Early genetic diagnosis enables tailored therapeutic interventions and family counseling
Therapeutic Strategies for Seizures in CDKL5 Deficiency Disorder
Pharmacologic Treatment Approaches
Seizures in CDKL5 deficiency are notoriously pharmacoresistant. Treatment regimens typically include:
- Vigabatrin (particularly for infantile spasms)
- Valproate, topiramate, and levetiracetam
- Steroids or ACTH during early infantile spasms
- Clobazam and rufinamide in Lennox-Gastaut-like presentations
Non-Pharmacologic Interventions
- Ketogenic diet: May provide seizure control in selected patients with drug-resistant epilepsy
- Vagus nerve stimulation (VNS): Considered in refractory cases
- Responsive neurostimulation (RNS): Under investigation
Investigational Therapies and Research Directions
Gene and Protein Replacement Approaches
Recent preclinical studies aim to:
- Restore CDKL5 protein function through gene therapy
- Explore mRNA-based therapies targeting mutant alleles
- Use CRISPR-Cas9 for gene correction in model systems
CDKL5 Deficiency-Specific Antiepileptic Trials
Clinical trials are exploring drugs such as ganaxolone, a neurosteroid modulating GABA-A receptors, which has shown promise in CDD-related seizures.
Prognosis and Long-Term Outlook
Despite aggressive treatment, most children continue to experience persistent seizures. The disorder also leads to:
- Severe intellectual disability
- Motor dysfunction (e.g., dystonia, spasticity)
- Visual impairment
- Autonomic instability
Prognosis is primarily determined by seizure control, early therapeutic intervention, and supportive care for comorbidities.
Multidisciplinary Management of CDKL5-Associated Seizures
Comprehensive Care Models
Optimal management requires collaboration between:
- Neurologists specializing in epilepsy genetics
- Physiatrists and therapists for motor and developmental support
- Ophthalmologists for cortical visual impairment
- Palliative care in advanced or refractory cases
Family and Psychosocial Support
Given the complexity of care, robust psychosocial services, including genetic counseling and caregiver mental health support, are essential.
Seizures in CDKL5 deficiency disorder represent a severe, early-onset, and pharmaco-resistant form of epilepsy demanding timely diagnosis and a multidisciplinary care approach. While current treatments offer limited seizure control, ongoing research into gene-targeted therapies provides hope for future precision-based interventions. Understanding the epileptogenic mechanisms and implementing integrated care pathways remain critical in improving outcomes for individuals with CDKL5-related epileptic encephalopathy.