Rhabdomyosarcoma (RMS) is a malignant tumor originating from skeletal muscle progenitors. It predominantly affects children and adolescents but can also occur in adults. As the most common soft tissue sarcoma in the pediatric population, RMS presents unique challenges in diagnosis and management.
Epidemiology and Risk Factors
RMS accounts for approximately 5% of all childhood cancers and about 50% of soft tissue sarcomas in children . The incidence is highest in children under 10 years of age, with a slight male predominance. While the exact cause remains unclear, certain genetic conditions, such as Li-Fraumeni syndrome and neurofibromatosis type 1, have been associated with an increased risk of developing RMS .
Histological Subtypes
RMS is classified into several histological subtypes, each with distinct clinical and prognostic implications:
- Embryonal RMS (ERMS): The most common subtype, typically affecting younger children and often arising in the head and neck region or genitourinary tract.
- Alveolar RMS (ARMS): More aggressive and frequently found in adolescents and young adults, commonly occurring in the extremities.
- Pleomorphic RMS: Rare and primarily seen in adults, characterized by a diverse cellular appearance.
- Spindle cell/sclerosing RMS: A rare variant with a better prognosis, often affecting paratesticular regions in infants and young children.
Clinical Presentation
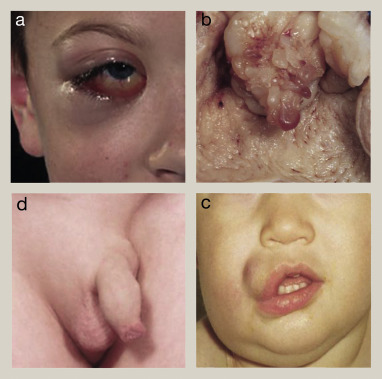
Symptoms of RMS vary depending on tumor location but may include:
- A rapidly growing, painless mass
- Obstructive symptoms (e.g., difficulty urinating or defecating)
- Proptosis or visual disturbances when located in the orbit
- Nasal obstruction or discharge for tumors in the nasal cavity
- Vaginal or testicular masses in genitourinary involvement
Early detection is crucial for optimal outcomes.
Diagnostic Evaluation
A thorough diagnostic workup is essential for accurate staging and treatment planning:
- Imaging Studies: MRI is preferred for local tumor assessment, while CT scans evaluate chest involvement. PET scans may detect metastatic disease.
- Biopsy: Core needle or surgical biopsy provides tissue for histological and molecular analysis.
- Bone Marrow Aspiration: Performed to assess marrow involvement, particularly in ARMS.
- Lumbar Puncture: Indicated if parameningeal involvement is suspected.
Staging and Risk Stratification
Staging combines tumor size, nodal involvement, and metastasis presence:
- Stage 1: Localized disease in favorable sites
- Stage 2: Localized disease in unfavorable sites, tumor ≤5 cm
- Stage 3: Localized disease in unfavorable sites, tumor >5 cm or regional nodal involvement
- Stage 4: Distant metastases present
Risk groups (low, intermediate, high) are determined based on stage, histology, and surgical resectability, guiding therapy intensity.
Treatment Modalities
Management of RMS requires a multidisciplinary approach:
- Surgery: Complete resection with negative margins is ideal. However, tumor location may limit surgical options.
- Chemotherapy: Standard regimens include vincristine, actinomycin D, and cyclophosphamide (VAC).
- Radiation Therapy: Employed for residual disease post-surgery or in unresectable tumors.
- Targeted Therapy: Investigational agents targeting specific molecular pathways are under study.
Prognosis
Prognostic factors influencing outcomes include:
- Age: Younger patients generally have better outcomes.
- Tumor Size and Location: Smaller tumors in favorable sites correlate with improved survival.
- Histological Subtype: ERMS has a better prognosis compared to ARMS.
- Metastatic Disease: Presence of metastases at diagnosis worsens prognosis.
Five-year survival rates vary:
- Low-risk group: >90%
- Intermediate-risk group: 70–80%
- High-risk group: <30%
Follow-Up and Survivorship
Long-term follow-up is essential to monitor for recurrence and manage late effects of therapy:
- Regular Imaging: Scheduled scans to detect recurrence
- Endocrine Evaluation: Assess growth and hormonal function, especially in children
- Psychosocial Support: Address emotional and developmental needs
- Rehabilitation Services: Physical and occupational therapy to improve function
Rhabdomyosarcoma, though rare, requires prompt recognition and a coordinated treatment strategy to optimize outcomes. Advances in multimodal therapy have improved survival rates, particularly in low and intermediate-risk groups. Ongoing research into targeted therapies and molecular characterization holds promise for further enhancing patient care.