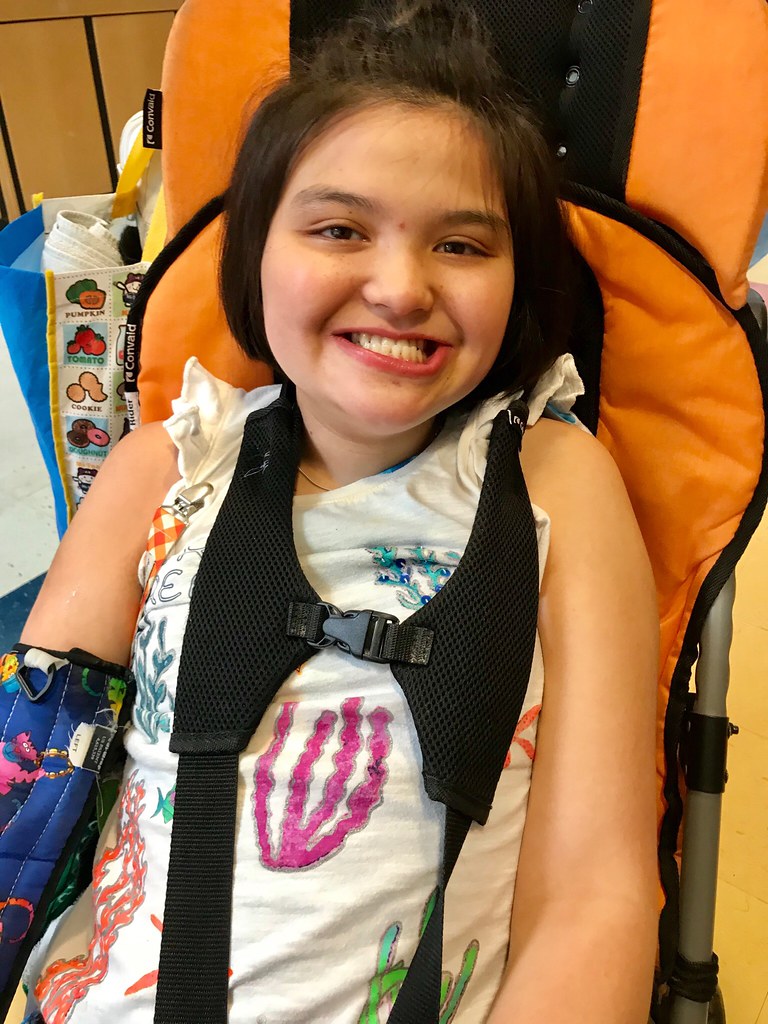
Rett syndrome is a rare, progressive neurodevelopmental disorder that predominantly affects females and leads to severe cognitive, motor, and communicative impairments. Caused by mutations in the MECP2 gene on the X chromosome, Rett syndrome manifests in early childhood and follows a characteristic clinical course with stages of regression. Though often misdiagnosed as autism or cerebral palsy, Rett syndrome is distinct in its genetic origin and specific progression pattern.
Genetic Basis of Rett Syndrome: The Role of MECP2
The MECP2 Gene and Its Function
The MECP2 gene (methyl CpG binding protein 2) plays a critical role in brain development and synaptic function. It regulates gene expression by binding to methylated DNA, thereby modulating neuronal maturation and plasticity.
Pathogenesis
Mutations in MECP2 disrupt neuronal communication, leading to:
- Impaired synaptic formation and maintenance
- Altered neurotransmitter activity
- Progressive loss of acquired skills
Approximately 95% of individuals with classic Rett syndrome have a pathogenic mutation in MECP2.
Epidemiology and Inheritance
- Incidence: ~1 in 10,000 to 15,000 live female births globally
- Sex Distribution: Primarily affects females; males with MECP2 mutations typically have severe neonatal encephalopathy and rarely survive infancy
- Inheritance: Most cases are sporadic, arising from de novo mutations; familial inheritance is extremely rare
Clinical Features and Key Symptoms of Rett Syndrome
Rett syndrome is characterized by normal early growth followed by regression and emergence of hallmark symptoms.
Early Signs (6 to 18 months)
- Loss of purposeful hand skills
- Delayed motor development
- Reduced eye contact
- Social withdrawal
Classic Symptoms
- Hand-wringing or stereotypic hand movements
- Loss of speech
- Gait abnormalities
- Seizures (in ~80%)
- Breathing irregularities (hyperventilation, apnea)
- Growth retardation (especially head circumference)
Stages of Rett Syndrome
Stage I: Early Onset (6–18 months)
- Subtle developmental delays
- Poor muscle tone
- Decreased eye contact
Stage II: Rapid Destructive Stage (1–4 years)
- Rapid loss of acquired skills
- Emergence of stereotypic hand movements
- Seizures may begin
Stage III: Plateau or Pseudo-Stationary Stage (2–10 years)
- Symptoms stabilize
- Improved behavior and communication
- Motor dysfunction continues
Stage IV: Late Motor Deterioration (10+ years)
- Muscle weakness, scoliosis, and mobility loss
- Cognitive and communication skills usually preserved from further decline
Differential Diagnosis and Related Conditions
Rett syndrome must be differentiated from conditions with overlapping features, such as:
- Autism spectrum disorder (ASD)
- Cerebral palsy
- Childhood disintegrative disorder
- Non-verbal developmental delay
MECP2 genetic testing is essential to confirm diagnosis.
Diagnostic Criteria and Genetic Testing
Clinical Diagnosis
Based on revised consensus criteria, diagnosis involves:
- Loss of purposeful hand skills
- Loss of spoken language
- Gait abnormalities
- Stereotypic hand movements
Confirmatory Testing
- MECP2 gene sequencing
- Chromosomal microarray (to rule out deletions)
- EEG and MRI for brain abnormalities
Early diagnosis allows timely intervention and management.
Treatment and Management Strategies
Rett syndrome has no cure; treatment focuses on symptom relief and supportive care.
Multidisciplinary Approach
- Neurologists: Manage seizures and sleep issues
- Physical/Occupational Therapists: Improve mobility and daily functioning
- Speech Therapists: Assist with non-verbal communication
- Nutritionists: Address feeding difficulties and malnutrition
Medications
- Anticonvulsants for seizures
- Muscle relaxants for spasticity
- Proton pump inhibitors for gastroesophageal reflux
Assistive Devices
- Gait trainers
- Adaptive communication tools
- Orthopedic supports
Emerging Therapies and Research Advances
Gene Therapy
Efforts to reactivate or replace MECP2 are ongoing, with promising results in animal models.
RNA Editing and Protein Modulation
Targeting downstream pathways affected by MECP2 mutation may offer alternative treatment options.
Clinical Trials
Recent trials include:
- Trofinetide: A synthetic analog of IGF-1 showing benefits in cognition and behavior
- Ketamine: Investigated for its neuroprotective effects
Long-term safety and efficacy remain under investigation.
Prognosis and Quality of Life
Life expectancy varies, but many individuals live into adulthood with appropriate care. Quality of life is closely linked to:
- Access to early intervention
- Seizure control
- Mobility preservation
- Supportive family and care networks
Ongoing support services are critical to optimize health and development.
Frequently Asked Questions:
What causes Rett syndrome?
A mutation in the MECP2 gene on the X chromosome disrupts brain development, leading to Rett syndrome.
Is Rett syndrome hereditary?
Most cases are not inherited but occur due to spontaneous mutations.
Can boys have Rett syndrome?
Rarely. It is typically lethal in males unless they have a less severe MECP2 mutation or an extra X chromosome.
Is there a cure for Rett syndrome?
Currently, there is no cure, but research into gene therapy and targeted treatments is ongoing.
What therapies help manage Rett syndrome?
Physical, occupational, and speech therapies alongside medication can significantly improve quality of life.
Rett syndrome remains one of the most complex pediatric neurological disorders. Although incurable, advances in genetic research and supportive multidisciplinary care are transforming the outlook for affected individuals. Early diagnosis, vigilant medical care, and dedicated family support systems are key to managing the challenges of Rett syndrome and enhancing the lives of those impacted.