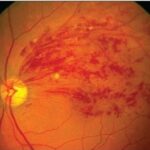
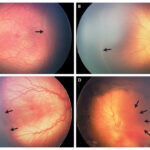
Retinoblastoma is a rare but aggressive malignant tumor that originates in the retina, primarily affecting infants and young children. It represents the most common primary intraocular cancer in childhood and demands swift intervention to preserve both vision and life. Early diagnosis, genetic counseling, and targeted treatment have significantly improved outcomes in recent decades.
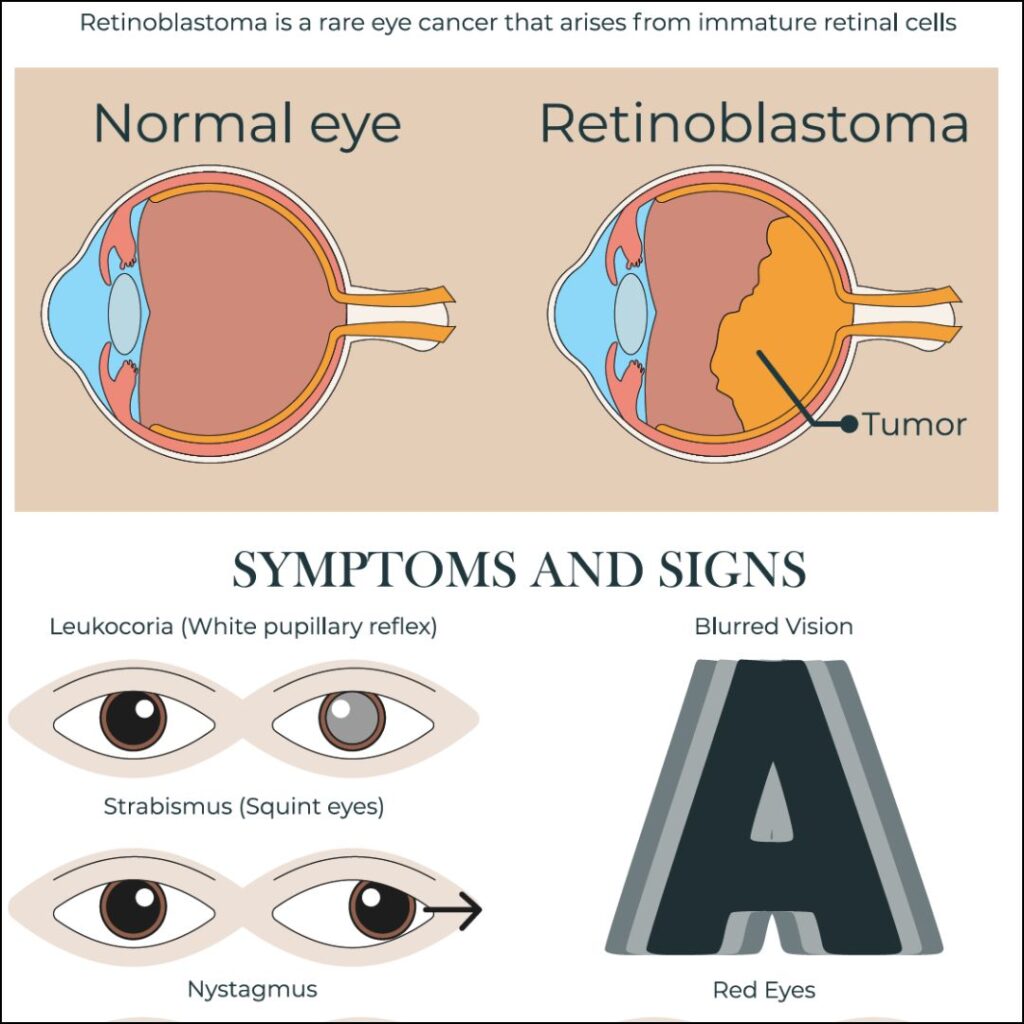
Genetic Origins and Classification
Retinoblastoma arises due to mutations in the RB1 tumor suppressor gene located on chromosome 13q14. This gene plays a critical role in regulating cell division. When both alleles of the RB1 gene are inactivated, retinal cells begin to proliferate uncontrollably, forming tumors.
Classification
Retinoblastoma is classified as:
- Heritable (Germline) Retinoblastoma
Accounts for approximately 40% of cases. It may be bilateral or multifocal and carries a higher risk of secondary cancers. - Non-heritable (Sporadic) Retinoblastoma
Typically unilateral with no family history and limited to one tumor.
Early Signs and Clinical Features
Retinoblastoma often presents subtly, and parents are usually the first to notice abnormalities.
Common Signs
- Leukocoria: A white pupillary reflex (“cat’s eye reflex”) observed in photographs
- Strabismus: Misalignment of the eyes due to impaired vision
- Poor visual tracking or fixation in infants
- Red, painful eye in advanced cases
Disease Progression
Untreated retinoblastoma can extend beyond the retina, invading the optic nerve, brain, or metastasizing to distant organs. Early intervention is essential to prevent extraocular spread.
Diagnostic Approach
Diagnosis is based on clinical evaluation and advanced imaging modalities, supported by genetic testing in suspected hereditary cases.
Ophthalmologic Examination
- Dilated fundus examination under anesthesia
- Indirect ophthalmoscopy to assess tumor size, location, and number
Imaging Studies
- Ocular ultrasound (B-scan): Detects calcification within the tumor
- MRI of brain and orbit: Evaluates optic nerve involvement and intracranial extension
- CT scan is generally avoided due to radiation risk, especially in heritable cases
Genetic Testing
Genetic analysis of RB1 gene mutations assists in confirming heritable retinoblastoma, identifying at-risk family members, and tailoring surveillance strategies.
Staging and International Classification
The International Intraocular Retinoblastoma Classification (IIRC) system stratifies intraocular tumors into five groups (A to E) based on size, location, and complications such as vitreous or subretinal seeding.
| Group | Description |
|---|---|
| A | Small tumors away from fovea and optic disc |
| B | Larger or closer tumors without seeding |
| C | Localized vitreous or subretinal seeding |
| D | Diffuse seeding with minimal retinal function |
| E | Extensive disease with poor visual prognosis |
Treatment Modalities
Management is individualized based on laterality, extent, vision potential, and systemic health.
Conservative Therapies (Eye-Sparing)
- Systemic Chemotherapy (Vincristine, Etoposide, Carboplatin): For tumor reduction and control
- Intra-arterial Chemotherapy (IAC): Direct delivery of chemotherapy to the ophthalmic artery
- Intravitreal Chemotherapy: Injection into the vitreous for treating seeding
- Laser Photocoagulation & Cryotherapy: Local tumor control in small, peripheral lesions
- Plaque Brachytherapy: Targeted radiation for localized tumors
Surgical Intervention
- Enucleation: Surgical removal of the eye is considered for advanced unilateral disease or when vision cannot be preserved. It prevents metastatic risk.
Radiation Therapy
- External Beam Radiotherapy (EBRT): Used selectively due to long-term risks including facial bone growth restriction and secondary malignancies in heritable cases.
Prognosis and Outcomes
With modern multidisciplinary care, survival rates exceed 95% in high-income countries. Factors influencing prognosis include:
- Early detection
- Unilateral vs. bilateral presentation
- Presence of high-risk features (e.g., optic nerve invasion)
- Accessibility to specialized care
Vision preservation is possible in many cases with eye-sparing treatments, particularly when diagnosed early.
Long-Term Follow-Up and Secondary Cancers
Children with heritable retinoblastoma require lifelong surveillance for:
- Second primary malignancies such as osteosarcoma, soft tissue sarcoma, or melanoma
- Psychosocial support due to visual impairment or cosmetic consequences
- Prosthetic eye care following enucleation
Genetic counseling for families is essential to guide future reproductive decisions and to screen siblings.
Preventive Strategies and Awareness
Although retinoblastoma itself cannot be prevented, early diagnosis can be life-saving. Preventive strategies include:
- Newborn red reflex screening by pediatricians
- Regular eye exams in at-risk children
- Genetic counseling and testing in families with history
- Public awareness campaigns to educate on signs like leukocoria
Frequently Asked Questions:
What age does retinoblastoma usually appear?
Most cases present before age 5, with a peak incidence between 18–24 months.
Can both eyes be affected?
Yes. Bilateral involvement is more common in heritable retinoblastoma.
Is retinoblastoma curable?
Yes. With prompt diagnosis and treatment, the survival rate is over 95% in developed countries.
What causes retinoblastoma?
Mutations in the RB1 gene, either inherited or sporadic, lead to uncontrolled cell growth in the retina.
How is it detected early?
White reflex in photos (leukocoria), eye misalignment, and routine red reflex screening help with early detection.
Is enucleation always necessary?
No. Many cases can be treated with chemotherapy or localized therapies to preserve the eye and vision.