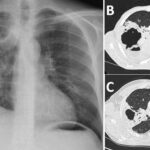
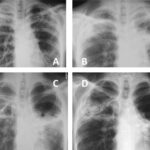
Pulmonary arterial hypertension (PAH) is a rare but severe condition characterized by elevated blood pressure in the pulmonary arteries, the blood vessels that carry blood from the heart to the lungs. PAH places excessive strain on the right side of the heart and can lead to heart failure if not diagnosed and treated early. The condition often progresses slowly, making it challenging to identify in its early stages. This article delves into the causes, symptoms, diagnosis, treatment options, and management strategies for pulmonary arterial hypertension to help improve patient outcomes.
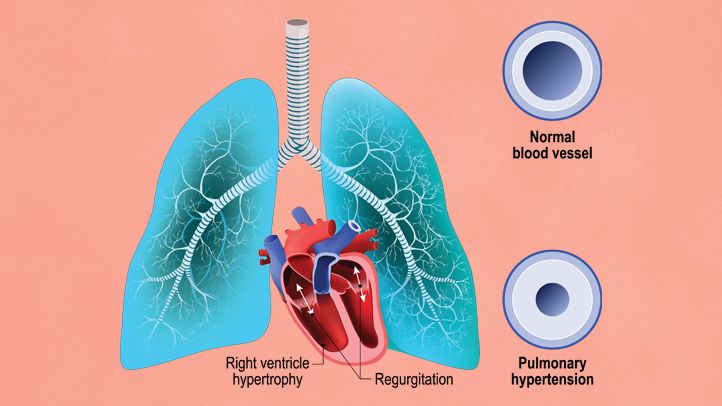
Causes and Risk Factors of Pulmonary Arterial Hypertension
PAH results from an increase in pulmonary artery pressure, which can occur due to a variety of underlying causes. In many cases, the cause is not entirely clear, but several risk factors have been identified.
1. Idiopathic Pulmonary Arterial Hypertension (IPAH)
In idiopathic cases, the cause of PAH is unknown. This form of PAH occurs without any identifiable predisposing condition or disease, making it particularly difficult to predict or prevent.
2. Heritable Pulmonary Arterial Hypertension (HPAH)
In some individuals, PAH is inherited due to mutations in specific genes, including the BMPR2 gene, which plays a role in the regulation of blood vessel growth and function. Genetic mutations can lead to abnormal blood vessel function and the development of PAH.
3. Associated Pulmonary Arterial Hypertension
PAH is also associated with several underlying conditions, including:
- Connective tissue diseases: Autoimmune diseases such as scleroderma, lupus, and rheumatoid arthritis can contribute to the development of PAH.
- Congenital heart defects: Individuals with certain heart conditions present from birth, such as atrial septal defects, are at a higher risk of developing PAH.
- Chronic lung diseases: Conditions such as chronic obstructive pulmonary disease (COPD) and interstitial lung disease can increase the likelihood of developing PAH due to the strain they place on the lungs and pulmonary arteries.
- HIV infection: The human immunodeficiency virus (HIV) can cause vascular changes, leading to an increased risk of developing PAH.
4. Other Risk Factors
Other factors contributing to PAH include:
- Obesity and lifestyle: Being overweight and sedentary can increase the risk of developing PAH.
- Sleep apnea: Conditions that interfere with normal breathing during sleep, such as obstructive sleep apnea, can place added pressure on the pulmonary arteries.
- Use of certain drugs: Some drugs, including appetite suppressants and certain chemotherapy agents, have been linked to the development of PAH.
Symptoms of Pulmonary Arterial Hypertension
The symptoms of PAH can be subtle at first and may mimic those of other conditions, which often leads to a delayed diagnosis. As the disease progresses, symptoms tend to worsen, and patients may experience:
1. Shortness of Breath (Dyspnea)
Difficulty breathing, especially during physical exertion, is one of the most common symptoms of PAH. As the pressure in the pulmonary arteries rises, the heart struggles to pump blood efficiently, leading to oxygenation problems in the lungs.
2. Fatigue
Persistent fatigue, even with minimal activity, is often reported by patients with PAH. The heart’s decreased ability to pump blood results in less oxygen being delivered to the body’s tissues, causing exhaustion.
3. Chest Pain
Patients with PAH may experience chest discomfort, which is typically related to the strain placed on the heart as it works harder to pump blood through narrowed pulmonary arteries.
4. Swelling (Edema)
Edema, particularly in the ankles, legs, and abdomen, is a common symptom. This occurs due to the heart’s inability to effectively pump blood, leading to fluid retention in various parts of the body.
5. Dizziness or Fainting
Due to reduced oxygen supply to the brain, individuals with PAH may experience dizziness, lightheadedness, or fainting, particularly during physical exertion or when standing up quickly.
6. Cyanosis
In severe cases, a bluish tint to the lips or skin (cyanosis) may develop due to insufficient oxygen levels in the blood.
Diagnosing Pulmonary Arterial Hypertension
The diagnosis of PAH requires a thorough evaluation that includes clinical assessment, imaging studies, and tests to measure the pressures within the pulmonary arteries.
1. Physical Examination and Medical History
During a physical examination, healthcare providers will assess for signs of right heart failure, such as swelling in the legs or abnormal heart sounds. A detailed medical history will help identify any risk factors or conditions associated with PAH.
2. Echocardiogram
An echocardiogram, which uses sound waves to create images of the heart, is a key diagnostic tool for evaluating heart function and estimating pulmonary artery pressure. While it does not provide a definitive diagnosis of PAH, it can suggest the presence of elevated pressure.
3. Right Heart Catheterization
The gold standard for diagnosing PAH is right heart catheterization. This procedure involves threading a catheter through a vein into the heart to directly measure the pressure in the pulmonary arteries. This test is essential to confirm PAH and differentiate it from other forms of pulmonary hypertension.
4. Pulmonary Function Tests (PFTs)
Pulmonary function tests assess how well the lungs are functioning by measuring the volume of air the lungs can hold and how effectively they transfer oxygen into the bloodstream.
5. Chest X-ray and CT Scan
Imaging studies such as a chest X-ray and CT scan provide valuable information about the size and condition of the heart and lungs. A CT scan is particularly useful for detecting signs of pulmonary disease and ruling out other causes of elevated pulmonary artery pressure.
Treatment and Management of Pulmonary Arterial Hypertension
While there is no cure for PAH, effective management can improve quality of life and help slow disease progression. Treatment strategies typically include medications, lifestyle changes, and in some cases, surgical interventions.
1. Medications
Several classes of medications are used to treat PAH, aiming to relax the pulmonary arteries and reduce blood pressure:
- Endothelin Receptor Antagonists (ERA): These medications, such as bosentan and ambrisentan, block the effects of endothelin, a molecule that constricts blood vessels.
- Phosphodiesterase-5 Inhibitors (PDE5 Inhibitors): Drugs like sildenafil and tadalafil help dilate blood vessels by blocking the enzyme phosphodiesterase-5, improving blood flow to the lungs.
- Prostacyclin Analogs: These medications, such as epoprostenol and treprostinil, mimic the effects of prostacyclin, a natural compound that dilates blood vessels and reduces platelet aggregation.
- Soluble Guanylate Cyclase Stimulators: Riociguat is a medication that helps relax and dilate the pulmonary arteries.
2. Oxygen Therapy
In cases where blood oxygen levels are insufficient, oxygen therapy may be prescribed to help improve oxygenation and reduce strain on the heart.
3. Surgical Intervention
In severe cases, surgery may be necessary to improve the quality of life or extend survival. Options include:
- Lung transplantation: For patients with end-stage PAH who do not respond to other treatments, lung transplantation may be considered.
- Atrial septostomy: In cases where there is severe right heart failure, an atrial septostomy may be performed to create a hole between the heart’s atria, reducing pressure in the lungs.
4. Lifestyle Changes
Lifestyle modifications, such as maintaining a healthy weight, avoiding excessive physical exertion, and managing stress, can help reduce the burden of symptoms. Patients are also advised to avoid high altitudes and ensure that they are properly vaccinated to prevent respiratory infections.
Prognosis of Pulmonary Arterial Hypertension
The prognosis for patients with PAH varies based on the severity of the condition at the time of diagnosis, the underlying cause, and how well the patient responds to treatment. With early detection and appropriate management, many individuals with PAH can lead active lives. However, if left untreated, PAH can progress to right heart failure and be fatal.
Pulmonary arterial hypertension is a life-threatening condition that requires early diagnosis and appropriate treatment to prevent severe complications. By understanding the causes, symptoms, and available therapies for PAH, patients can work with healthcare providers to manage the disease effectively and improve their quality of life. Advances in pharmacological therapies and surgical interventions continue to offer hope for those affected by this complex condition.