Propionic acidemia (PA) is a rare but serious inherited metabolic disorder belonging to the group of organic acidemias. It results from a deficiency in the enzyme propionyl-CoA carboxylase (PCC), which is essential for breaking down certain proteins and fats. Without this enzyme, toxic levels of propionic acid accumulate in the body, leading to severe metabolic imbalances. Understanding the biochemical, clinical, and genetic dimensions of this disorder is critical for effective diagnosis, management, and support for affected individuals.
Pathophysiology and Biochemical Basis of Propionic Acidemia
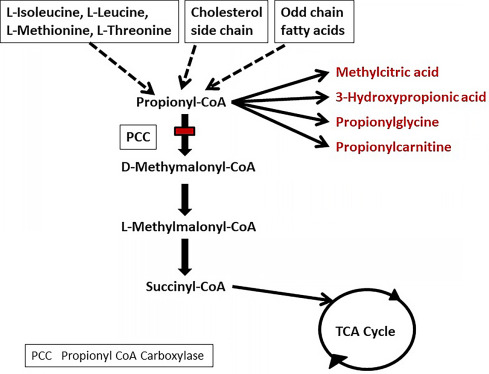
Enzyme Deficiency in PCC Activity
Propionic acidemia is caused by mutations in the PCCA or PCCB genes, which encode the two subunits of the propionyl-CoA carboxylase enzyme. This enzyme is responsible for converting propionyl-CoA into methylmalonyl-CoA, a critical step in the metabolism of branched-chain amino acids (isoleucine, valine, methionine, threonine), odd-chain fatty acids, and cholesterol side chains.
In PA, the defective enzyme leads to accumulation of toxic metabolic intermediates, primarily propionic acid, which impairs cellular functions, especially in the brain, heart, and liver.
Clinical Manifestations of Propionic Acidemia
Neonatal-Onset PA
In its most severe form, propionic acidemia manifests shortly after birth. Common symptoms include:
- Lethargy and poor feeding
- Vomiting and dehydration
- Hypotonia (reduced muscle tone)
- Metabolic acidosis with elevated anion gap
- Hyperammonemia (elevated blood ammonia)
- Seizures and coma in severe cases
These symptoms often present within the first few days of life and require immediate medical attention.
Late-Onset and Intermittent Forms
Some individuals with milder mutations may present later in infancy or childhood with:
- Developmental delays
- Growth retardation
- Chronic vomiting
- Movement disorders (e.g., dystonia)
- Cardiomyopathy or heart rhythm abnormalities
Metabolic decompensation can occur during stress, illness, or fasting, leading to life-threatening crises.
Diagnostic Approach for Propionic Acidemia
Newborn Screening and Laboratory Investigations
Propionic acidemia is included in newborn screening panels in many countries. Early detection allows for prompt intervention. Confirmatory diagnostic tests include:
- Urine organic acid analysis: Elevated propionic acid, 3-hydroxypropionic acid, and methylcitrate
- Plasma amino acids: Low glutamine, high glycine
- Blood acylcarnitine profile: Elevated propionylcarnitine (C3)
- Molecular genetic testing: Identification of mutations in PCCA or PCCB genes
Treatment and Long-Term Management Strategies
Acute Management
During metabolic crises, immediate treatment aims to reverse catabolism and reduce toxic metabolite levels. This involves:
- Intravenous glucose and lipids to provide non-protein calories
- Hemodialysis or peritoneal dialysis in severe cases with hyperammonemia
- Intravenous carnitine to enhance excretion of toxic intermediates
- Antibiotics like metronidazole to reduce propionate-producing gut bacteria
Chronic Management
Ongoing treatment aims to prevent decompensation and support normal development. Key components include:
- Protein-restricted diet: Limiting intake of propionyl-CoA-producing amino acids
- Special metabolic formula: Provides essential nutrients while avoiding toxic precursors
- Carnitine supplementation: Helps transport toxic acyl groups out of mitochondria
- Biotin supplementation: In rare responsive cases, supports residual enzyme activity
- Regular monitoring: Blood ammonia, acid-base status, growth parameters
Gene Therapy and Future Perspectives
Advances in Genetic Medicine
Research into gene therapy offers hope for more definitive treatments. Experimental approaches involve:
- Gene replacement therapy: Using viral vectors to deliver functional PCCA or PCCB genes
- mRNA-based therapies: Temporary replacement of deficient enzyme activity
- CRISPR gene editing: Direct correction of mutations at the genomic level
While these therapies are still under investigation, early trials in animal models show promise in reducing metabolic abnormalities and improving survival.
Prognosis and Quality of Life
The long-term outlook for individuals with propionic acidemia varies depending on the severity of enzyme deficiency, timeliness of diagnosis, and effectiveness of treatment. With early intervention and meticulous metabolic control:
- Many children achieve developmental milestones, albeit often with some delays
- Cardiac, neurologic, and hepatic complications remain challenges
- Multidisciplinary care, including metabolic specialists, neurologists, cardiologists, and dietitians, is essential for optimal outcomes
Genetic Counseling and Family Planning
As an autosomal recessive disorder, propionic acidemia carries a 25% recurrence risk in future pregnancies if both parents are carriers. Genetic counseling is crucial for affected families. Options include:
- Carrier testing for parents and relatives
- Prenatal diagnosis via chorionic villus sampling or amniocentesis
- Preimplantation genetic diagnosis (PGD) for families using in vitro fertilization
Propionic acidemia is a complex, life-threatening disorder that demands early recognition, aggressive management, and lifelong care. With advances in newborn screening, dietary therapy, and genetic research, the prognosis continues to improve. Continued awareness, clinical vigilance, and innovation in therapy are essential to ensure the best outcomes for patients with this rare condition.