Primary Immune Deficiency Disorder (PIDD) refers to a group of over 400 rare, chronic disorders in which part of the body’s immune system is either missing or functions improperly. These conditions are inherited genetic defects that compromise the immune system’s ability to defend against infections, malignancies, and autoimmune diseases. Individuals with PIDD are more susceptible to recurrent, severe, and sometimes life-threatening infections.
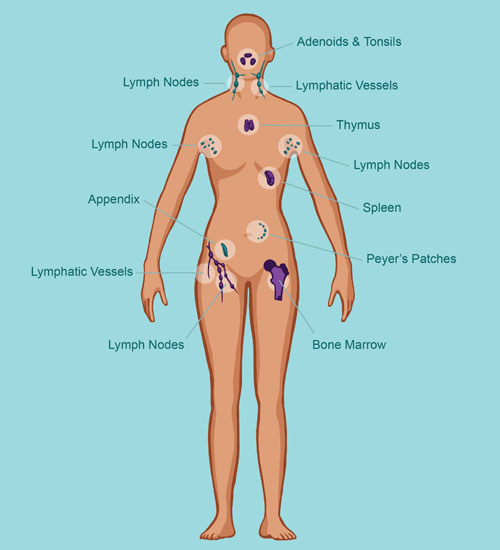
Classification of Primary Immune Deficiency Disorders
Major Categories of PIDD
PIDDs are categorized based on the specific components of the immune system affected:
- B-cell (antibody) deficiencies: e.g., X-linked agammaglobulinemia, Common Variable Immune Deficiency (CVID)
- T-cell deficiencies: e.g., DiGeorge syndrome
- Combined B- and T-cell deficiencies: e.g., Severe Combined Immunodeficiency (SCID)
- Phagocytic defects: e.g., Chronic Granulomatous Disease (CGD)
- Complement deficiencies: e.g., C3 deficiency, C1 esterase inhibitor deficiency
Genetic and Molecular Basis of PIDD
Inherited Mutations and Immune Dysfunction
PIDDs result from germline mutations in genes crucial for immune cell development and function. These mutations are often inherited in an autosomal recessive or X-linked pattern. For example:
- BTK gene mutations cause X-linked agammaglobulinemia by halting B-cell maturation.
- ADA or IL2RG mutations lead to SCID, severely affecting lymphocyte development.
The Role of Genetic Testing
Genomic sequencing and next-generation panels help confirm PIDD diagnoses, offering valuable insights for:
- Tailoring treatment
- Predicting disease progression
- Identifying suitable candidates for gene therapy
Clinical Manifestations of Primary Immune Deficiency
Frequent Infections
Patients often present with:
- Recurrent respiratory infections (sinusitis, bronchitis, pneumonia)
- Chronic ear infections
- Gastrointestinal infections and persistent diarrhea
- Skin abscesses or oral thrush
Autoimmune and Inflammatory Complications
Many individuals develop:
- Autoimmune cytopenias (e.g., ITP, hemolytic anemia)
- Rheumatologic symptoms resembling lupus or juvenile arthritis
- Enteropathy mimicking inflammatory bowel disease
Delayed Diagnosis and Misdiagnosis
Due to overlap with common illnesses, diagnosis is often delayed. Clinicians must maintain a high index of suspicion, especially in cases of:
- Infections requiring repeated antibiotics
- Failure to thrive in children
- Family history of early deaths or recurrent infections
Diagnostic Approach to Primary Immune Deficiency Disorders
Initial Laboratory Evaluation
- Complete blood count (CBC) with differential
- Serum immunoglobulin levels (IgG, IgA, IgM)
- Lymphocyte subset analysis by flow cytometry
Advanced Immunological Testing
- Specific antibody response to vaccines (e.g., tetanus, pneumococcus)
- Neutrophil function tests
- Complement assays
- Molecular genetic testing when PIDD is suspected based on phenotype
Long-Term Management of Primary Immune Deficiencies
Immunoglobulin Replacement Therapy
A cornerstone of treatment for B-cell deficiencies:
- Intravenous immunoglobulin (IVIG) or subcutaneous immunoglobulin (SCIG)
- Reduces frequency and severity of infections
- Requires lifelong administration in most cases
Hematopoietic Stem Cell Transplant (HSCT)
For severe combined immunodeficiencies and selected PIDDs:
- Allogeneic bone marrow transplantation can be curative
- Best outcomes with early diagnosis and HLA-matched donors
Emerging Gene Therapy
Revolutionary progress in gene editing has enabled:
- Autologous gene-corrected stem cell therapy (e.g., ADA-SCID)
- Trials show promise with long-term immune reconstitution and fewer infections
Supportive and Preventive Measures
- Antibiotic prophylaxis to prevent opportunistic infections
- Vaccinations (live vaccines are contraindicated in many PIDDs)
- Nutritional support and regular monitoring
- Pulmonary and gastrointestinal care to manage organ-specific complications
Living with PIDD: Patient Outlook and Quality of Life
Though PIDDs are chronic, early intervention and comprehensive care significantly improve outcomes. Children can attend school, adults can work and lead fulfilling lives with proper treatment adherence, regular follow-ups, and emotional support. Patient education and awareness are essential for recognizing early signs of complications and preventing serious infections.
Primary Immune Deficiency Disorders are complex yet manageable conditions. Timely diagnosis, precision treatment, and a multidisciplinary approach are essential to minimize morbidity and maximize quality of life. With ongoing advancements in immunology and genetics, the future holds promising therapies that may offer lasting cures and improved immune restoration.