Primary Hyperoxaluria Type I (PH1) is a rare autosomal recessive genetic disorder characterized by the overproduction of oxalate, a metabolic end product that the body cannot degrade. The excess oxalate combines with calcium to form calcium oxalate crystals, leading to nephrolithiasis, nephrocalcinosis, and ultimately end-stage kidney disease (ESKD) if left untreated.
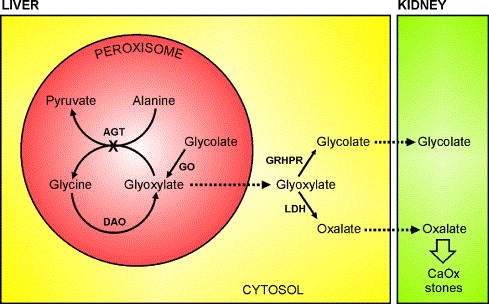
PH1 results from a deficiency or malfunction of the liver enzyme alanine-glyoxylate aminotransferase (AGT), encoded by the AGXT gene. This enzyme plays a pivotal role in glyoxylate detoxification within hepatic peroxisomes. The failure of this process leads to systemic oxalosis—a condition marked by oxalate accumulation in bones, eyes, blood vessels, and other organs.
Genetic and Molecular Basis
AGXT Gene and Enzyme Function
PH1 is caused by mutations in the AGXT gene located on chromosome 2q37.3. The AGXT gene encodes AGT, an enzyme responsible for converting glyoxylate to glycine. Over 150 mutations in AGXT have been identified, with G170R, F152I, and I244T being among the most common.
Mislocalization and Functional Deficiency
Some AGXT mutations lead to mislocalization of AGT from the peroxisomes to mitochondria, rendering the enzyme non-functional. Others directly reduce catalytic activity or expression levels, all contributing to oxalate accumulation.
Clinical Manifestations of PH1
Renal Symptoms
- Recurrent kidney stones (nephrolithiasis)
- Nephrocalcinosis (diffuse calcification of renal parenchyma)
- Progressive renal insufficiency
- Hematuria and flank pain
Systemic Oxalosis (in advanced stages)
- Bone pain and fractures
- Anemia and bone marrow suppression
- Retinal oxalate deposits causing visual impairment
- Cardiac conduction defects and arrhythmias
- Skin ulcers and systemic organ dysfunction
Age of Onset
PH1 can present in infancy, childhood, or adulthood, with the infantile form associated with rapid progression to kidney failure and poor prognosis if untreated.
Diagnostic Approach for Primary Hyperoxaluria Type I
Biochemical Testing
- 24-hour urinary oxalate excretion: Typically >1 mmol/1.73 m²/day (normal <0.5)
- Elevated urinary glycolate and L-glycerate may suggest PH1 and PH2, respectively
- Plasma oxalate levels in renal failure cases
Imaging Studies
- Renal ultrasound: Nephrocalcinosis and calculi
- Non-contrast CT: Superior for stone detection
Genetic Confirmation
- AGXT gene sequencing is definitive for PH1
- Prenatal diagnosis via chorionic villus sampling (CVS) or amniocentesis in families with known mutations
Liver Biopsy (rarely used now)
- Enzyme assay for AGT activity (invasive, largely replaced by genetic testing)
Management and Treatment of PH1
Conservative Measures
- High fluid intake: ≥3 L/m²/day to dilute urinary oxalate
- Potassium citrate or sodium bicarbonate: To alkalinize urine and inhibit crystal formation
- Pyridoxine (Vitamin B6): May reduce oxalate synthesis in patients with specific AGXT mutations (e.g., G170R)
Pharmacologic Advances
Lumasiran (Oxlumo™)
- RNA interference (RNAi) therapy targeting glycolate oxidase
- Reduces glyoxylate conversion to oxalate
- Approved by the FDA and EMA for all ages
- Administered via subcutaneous injection monthly for 3 months, then quarterly
Nedosiran
- Investigational RNAi therapy targeting LDHA (lactate dehydrogenase A)
- Reduces oxalate production systemically
- Currently under clinical evaluation
Dialysis
- Limited efficacy in oxalate clearance
- Often serves as a bridge to transplantation
- Requires both hemodialysis and peritoneal dialysis to optimize oxalate removal
Transplantation
Isolated Liver Transplant
- Cures metabolic defect by restoring AGT activity
- Suitable in patients with preserved renal function but high oxalate load
Combined Liver-Kidney Transplant
- Recommended in advanced kidney failure
- Addresses both enzymatic deficiency and renal damage
- Considered curative
Emerging Therapies and Research Directions
Gene Therapy
- Preclinical studies are exploring AGXT gene replacement and CRISPR-based gene correction
- Aims to provide permanent cure without transplantation
Enzyme Replacement and mRNA Therapies
- Engineered recombinant AGT enzymes or mRNA-based therapies may offer alternatives for direct enzyme restoration
Genetic Counseling and Family Screening
- As an autosomal recessive disorder, each sibling of an affected individual has a 25% risk of being affected
- Carrier testing and genetic counseling are crucial for family planning
- Newborn screening is under consideration in populations with higher carrier frequency
Prognosis and Long-Term Management
Factors Influencing Prognosis
- Age at diagnosis
- Presence of pyridoxine-responsive mutations
- Timing of intervention
- Access to transplantation or RNAi therapy
Monitoring Protocol
- Regular assessment of urinary oxalate and glycolate
- Renal imaging and function monitoring
- Annual screening for systemic oxalosis in advanced cases
Primary Hyperoxaluria Type I is a life-threatening but increasingly manageable condition thanks to early diagnosis, targeted therapies, and advances in genetic medicine. The introduction of RNAi-based treatments like Lumasiran and potential gene therapies represent a paradigm shift in PH1 care. With a multidisciplinary approach encompassing nephrology, hepatology, genetics, and urology, we can significantly improve patient outcomes and quality of life.