it is also known as congenital hypoplasminogenemia, is a rare autosomal recessive disorder characterized by a marked reduction in plasminogen activity, which leads to the abnormal accumulation of fibrin-rich pseudomembranes on mucous membranes and other tissues. The disease often manifests early in life and can affect various organs, with serious implications for ocular, respiratory, and genitourinary health.
Understanding Plasminogen Deficiency Type 1
Plasminogen is a glycoprotein synthesized in the liver, serving as the precursor to plasmin, a key enzyme involved in fibrinolysis—the breakdown of fibrin in blood clots. In type 1 plasminogen deficiency, there is a quantitative defect, meaning both the level and function of plasminogen are reduced. This impairs the body’s ability to degrade fibrin, resulting in excessive deposition and formation of ligneous lesions.
Genetic Basis and Pathophysiology of Type 1 Plasminogen Deficiency
Plasminogen deficiency type 1 is inherited in an autosomal recessive manner. The condition results from mutations in the PLG gene, located on chromosome 6q26. This gene encodes plasminogen, and its mutation leads to decreased synthesis or secretion of the protein.
Key Molecular Pathways:
This impaired fibrinolytic system leads to chronic inflammation, tissue scarring, and pseudomembrane formation, particularly on mucosal surfaces.
Clinical Manifestations and Symptoms
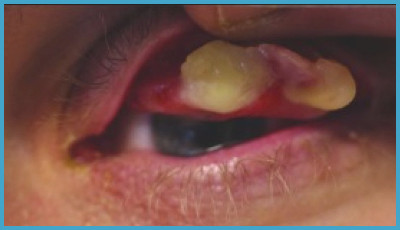
The hallmark symptom of type 1 plasminogen deficiency is ligneous conjunctivitis, a condition marked by thick, woody pseudomembranes on the conjunctiva of the eyes. However, symptoms can extend to other systems.
Ocular System
- Ligneous conjunctivitis
- Eyelid swelling
- Conjunctival hyperemia
- Vision impairment due to corneal involvement
Respiratory System
- Recurrent upper respiratory tract infections
- Formation of fibrinous lesions in the trachea and bronchi
- Airway obstruction in severe cases
Genitourinary System
- Ligneous lesions in the cervix or urethra
- Vaginal discharge or urinary retention
Gastrointestinal and Other Sites
- Gingival hypertrophy
- Fibrin deposits on gastrointestinal mucosa
- Wound healing complications
Diagnosis of Plasminogen Deficiency Type 1
A definitive diagnosis relies on laboratory assays and molecular genetic testing.
Diagnostic Methods:
- Plasminogen activity assays: Significantly reduced activity (<45% of normal)
- Plasminogen antigen level: Low concentration in type 1 (normal in type 2)
- Genetic testing: Identification of biallelic PLG gene mutations
- Histopathology: Presence of eosinophilic fibrin-rich pseudomembranes
Differential Diagnosis:
- Ligneous conjunctivitis from other etiologies
- Immunodeficiency disorders
- Autoimmune mucosal diseases
Treatment Strategies and Management
There is currently no curative therapy for type 1 plasminogen deficiency, but symptomatic and supportive treatments are available to control lesion development and improve quality of life.
1. Plasminogen Replacement Therapy
- Human plasma-derived plasminogen concentrate (Ryplazim): Approved by the FDA
- Used for systemic replacement therapy to reduce recurrence of lesions
2. Topical and Local Therapies
- Topical plasminogen drops: For ocular involvement
- Heparin and corticosteroid drops: Reduce inflammation
- Surgical removal of pseudomembranes: Often combined with adjunctive therapy
3. Supportive and Preventive Care
- Regular ophthalmologic follow-up
- Airway monitoring in patients with respiratory involvement
- Gynecological surveillance for female patients
- Avoidance of trauma to mucosal surfaces
4. Experimental and Emerging Therapies
- Gene therapy targeting PLG mutations (in preclinical stages)
- Recombinant plasminogen agents under investigation
- Anti-fibrinolytic balance regulators to manage deposition
Prognosis and Long-Term Outlook
Without treatment, plasminogen deficiency type 1 can result in progressive lesion development, organ damage, and significant morbidity. Early diagnosis and regular monitoring improve outcomes. Long-term replacement therapy and integrated care have shown promise in reducing recurrence and preserving organ function.
FAQs:
Q1: What causes plasminogen deficiency type 1?
It is caused by mutations in the PLG gene that result in a significant decrease in plasminogen production and function.
Q2: Is it a hereditary condition?
Yes, it is inherited in an autosomal recessive pattern, requiring two mutated copies of the gene.
Q3: What is the most common symptom?
Ligneous conjunctivitis, with thick, fibrin-rich pseudomembranes in the eye, is the most prevalent manifestation.
Q4: Can the disease be cured?
There is no cure, but plasminogen replacement and topical therapies can effectively manage symptoms.
Q5: How is it diagnosed?
Diagnosis involves plasminogen activity assays, antigen testing, genetic screening, and histopathology.
Plasminogen deficiency type 1 represents a complex, rare genetic disorder with profound implications for mucosal and systemic health. Through timely diagnosis, targeted therapies, and multidisciplinary care, affected individuals can experience significant symptom relief and improved quality of life. Advancements in plasminogen replacement therapies and genetic research offer promising prospects for more effective treatment and potential disease-modifying interventions in the future.