Pituitary-dependent Cushing’s disease is a specific subtype of Cushing’s syndrome caused by excessive secretion of adrenocorticotropic hormone (ACTH) from a pituitary adenoma, typically a microadenoma. This leads to chronic overstimulation of the adrenal glands and hypercortisolism, resulting in a wide array of systemic complications. Early recognition, accurate diagnosis, and appropriate treatment are crucial to improving patient outcomes and reducing morbidity.
Understanding the Pathophysiology of Pituitary-Dependent Cushing’s Disease
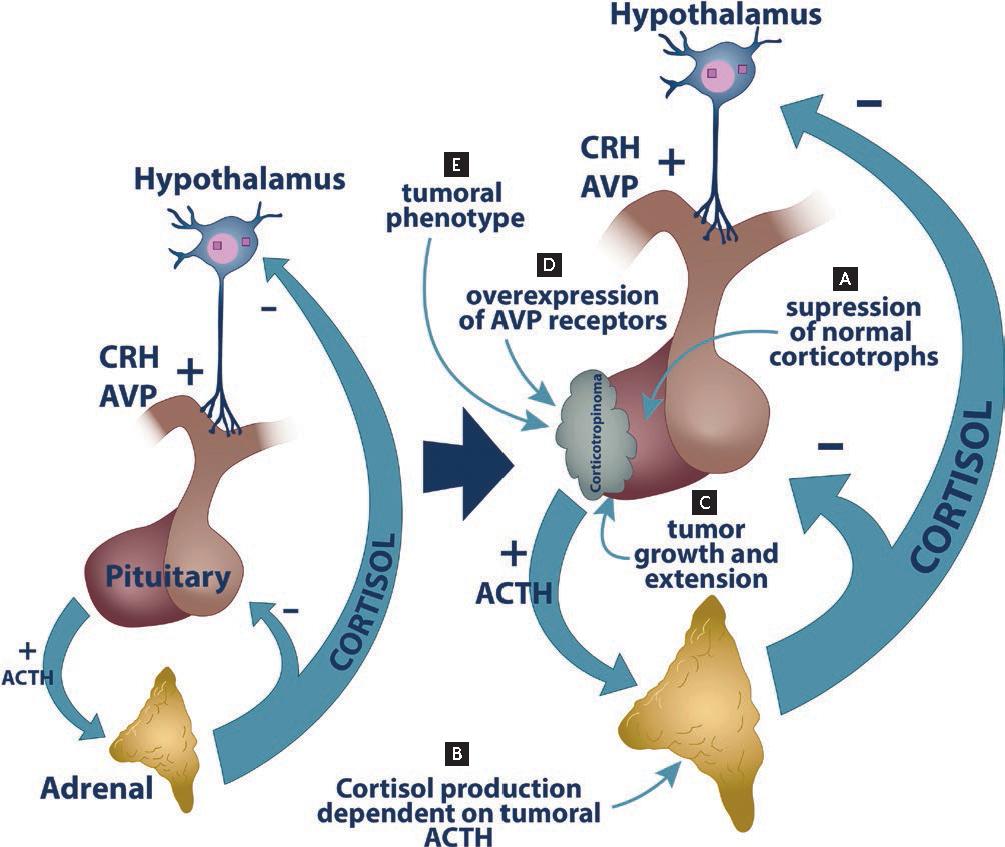
Cushing’s disease results from autonomous ACTH production by a benign pituitary tumor, disrupting the hypothalamic-pituitary-adrenal (HPA) axis. This leads to:
- Excessive cortisol secretion from the adrenal cortex.
- Loss of circadian rhythm of cortisol production.
- Negative feedback failure, allowing ACTH levels to remain elevated despite high cortisol levels.
Unlike ectopic ACTH syndrome or adrenal tumors, the pituitary etiology exhibits partial feedback suppression, a characteristic exploited in differential testing.
Clinical Features and Symptoms of Pituitary Cushing’s Disease
Patients with pituitary-dependent Cushing’s disease often present with insidious, progressive symptoms. The hallmark signs result from prolonged exposure to elevated cortisol levels:
- Central obesity (face, trunk, abdomen)
- “Moon face” and “buffalo hump”
- Facial plethora and thin skin
- Purple striae on the abdomen and thighs
- Proximal muscle weakness
- Easy bruising and poor wound healing
- Hypertension and glucose intolerance
- Menstrual irregularities in women
- Depression, anxiety, and cognitive dysfunction
- Osteoporosis and vertebral fractures
Pediatric cases often manifest with growth retardation and weight gain.
Diagnostic Evaluation of Pituitary-Dependent Cushing’s Disease
Accurate diagnosis involves confirming hypercortisolism, determining its ACTH dependency, and localizing the pituitary source. The work-up consists of biochemical tests and imaging studies.
Step 1: Confirm Hypercortisolism
At least two of the following tests are recommended:
- 24-hour urinary free cortisol (UFC): Measures unbound cortisol; values ≥2-3x upper limit suggest Cushing’s.
- Late-night salivary cortisol: Detects loss of diurnal rhythm.
- Low-dose dexamethasone suppression test (LDDST):
- 1 mg dexamethasone at 11 PM.
- Serum cortisol measured at 8 AM.
- Lack of suppression (cortisol >1.8 µg/dL) indicates hypercortisolism.
Step 2: Determine ACTH Dependency
- Plasma ACTH levels:
- ACTH >15 pg/mL: ACTH-dependent (pituitary or ectopic).
- ACTH <5 pg/mL: Suggests adrenal cause.
Step 3: Localize the ACTH Source
- High-dose dexamethasone suppression test (HDDST):
- 8 mg dexamethasone overnight or 2 mg q6h for 2 days.
- 50% suppression of cortisol suggests pituitary source.
- CRH stimulation test:
- Cortisol and ACTH rise ≥20% and ≥35% respectively supports pituitary etiology.
- MRI of the pituitary:
- Detects pituitary microadenomas (>70% cases).
- Sensitivity varies; up to 40% of tumors are <2 mm and may be missed.
- Inferior petrosal sinus sampling (IPSS):
- Gold standard for distinguishing pituitary from ectopic sources.
- Central-to-peripheral ACTH gradient >2 at baseline or >3 post-CRH confirms pituitary source.
Treatment Options for Pituitary Cushing’s Disease
1. Transsphenoidal Surgery (TSS)
First-line treatment targeting the ACTH-producing pituitary adenoma.
- Success rates: 70–90% for microadenomas.
- Lower remission in macroadenomas or invisible tumors.
- Requires experienced neurosurgical teams.
- Postoperative hypocortisolism often indicates remission.
2. Radiotherapy
- Indicated when surgery is unsuccessful or contraindicated.
- Conventional fractionated RT or stereotactic radiosurgery (e.g., Gamma Knife).
- Remission may take months to years.
3. Medical Therapy
Used when surgery is delayed, failed, or as a bridge to radiotherapy.
Classes of medications:
| Drug | Class | Mechanism |
|---|---|---|
| Ketoconazole | Adrenal enzyme inhibitor | Blocks steroidogenesis |
| Metyrapone | Adrenal enzyme inhibitor | Inhibits 11β-hydroxylase |
| Osilodrostat | Adrenal enzyme inhibitor | Inhibits cortisol synthesis |
| Pasireotide | Somatostatin analog | Suppresses ACTH secretion |
| Cabergoline | Dopamine agonist | Inhibits ACTH in some tumors |
| Mifepristone | Glucocorticoid receptor blocker | Antagonizes cortisol effects |
4. Bilateral Adrenalectomy
Reserved for refractory cases or when immediate cortisol reduction is necessary.
- Requires lifelong steroid replacement.
- Risk of Nelson’s syndrome (aggressive pituitary tumor growth due to loss of feedback inhibition).
Prognosis and Long-Term Follow-Up
- Recurrence occurs in up to 25% of cases post-surgery; hence, regular monitoring is essential.
- Long-term complications: cardiovascular disease, osteoporosis, neuropsychiatric symptoms.
- Annual screening of UFC, ACTH, and pituitary MRI is recommended post-remission.
Pituitary-dependent Cushing’s disease represents a challenging endocrine disorder with significant systemic impact. A meticulous and structured diagnostic approach, coupled with timely surgical intervention and appropriate adjuvant therapy, remains the cornerstone of effective management. Long-term surveillance is vital due to the potential for recurrence and persistent comorbidities, underscoring the importance of multidisciplinary endocrine care.