Pheochromocytoma is a rare catecholamine-producing tumor that arises from the chromaffin cells of the adrenal medulla. It is characterized by excessive secretion of epinephrine, norepinephrine, and, less commonly, dopamine, leading to potentially life-threatening hypertension and multisystemic complications. Early recognition, accurate diagnosis, and prompt intervention are critical for favorable outcomes.
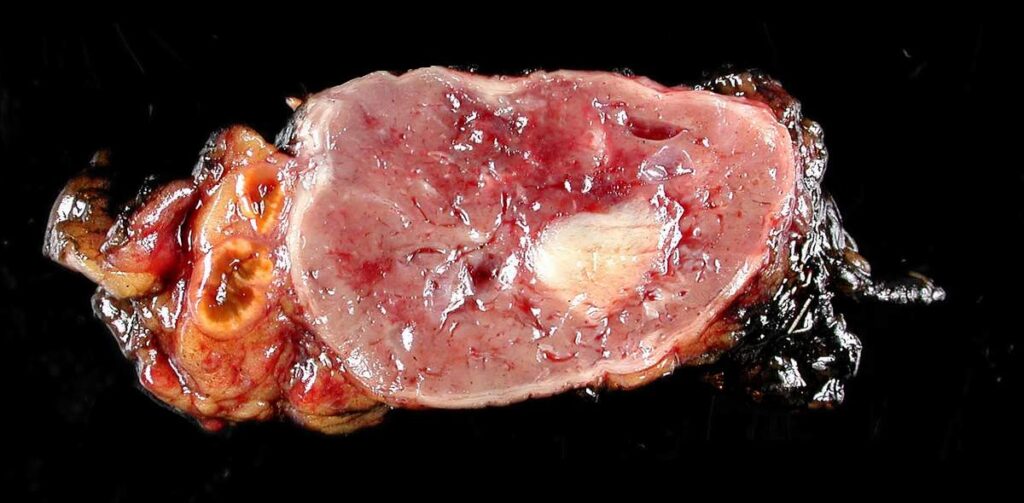
Origin and Pathophysiology of Pheochromocytoma
Pheochromocytomas originate from the chromaffin cells of the adrenal medulla and, in some cases, from extra-adrenal paraganglia (termed paragangliomas when found outside the adrenal glands). These tumors result in episodic or sustained catecholamine secretion, which severely disrupts cardiovascular homeostasis.
Epidemiology and Risk Factors
- Incidence: 2–8 cases per million people annually
- Peak age: 30–50 years
- Gender: Equal distribution
- Hereditary forms: Account for up to 40% of cases
Genetic Syndromes Associated with Pheochromocytoma
- Multiple Endocrine Neoplasia type 2 (MEN 2A/2B)
- Von Hippel-Lindau disease
- Neurofibromatosis type 1
- Succinate dehydrogenase (SDHx) mutations
Classic Symptoms of Pheochromocytoma
Pheochromocytomas are known for their hallmark “5 P’s” symptoms:
- Pressure (Hypertension) – sustained or paroxysmal
- Pain (Headache) – often pounding
- Perspiration – profuse, episodic
- Palpitations – rapid heartbeat or arrhythmias
- Pallor – due to peripheral vasoconstriction
Additional symptoms may include:
- Tremors
- Anxiety or panic attacks
- Nausea and vomiting
- Weight loss
- Hyperglycemia
Hypertensive crises may occur during surgery, trauma, or childbirth if undiagnosed.
Diagnostic Approach to Pheochromocytoma
Biochemical Testing
Accurate biochemical testing is the cornerstone of diagnosis.
- Plasma free metanephrines: Highly sensitive
- 24-hour urinary fractionated metanephrines and catecholamines: Useful for confirmation
- Clonidine suppression test: For equivocal cases
Imaging Studies
After biochemical confirmation, localization of the tumor is essential.
- CT Scan or MRI of the abdomen: Preferred initial modality
- MIBG scintigraphy (metaiodobenzylguanidine): For functional imaging
- PET scans (e.g., 18F-FDOPA, 18F-FDG): Especially in metastatic or hereditary cases
Differential Diagnosis
It is essential to distinguish pheochromocytoma from conditions with overlapping symptoms, including:
- Hyperthyroidism
- Panic disorder
- Carcinoid syndrome
- Labile essential hypertension
- Drug-induced sympathetic stimulation (e.g., cocaine)
Preoperative Management and Stabilization
The primary goal before surgery is to prevent intraoperative hypertensive crises.
Alpha-Blockade
- Phenoxybenzamine: Non-selective, long-acting alpha-blocker
- Doxazosin or Prazosin: Selective alpha-1 blockers with fewer side effects
Beta-Blockade
- Only after adequate alpha-blockade
- Prevents unopposed alpha-adrenergic stimulation
- Propranolol or Atenolol commonly used
Volume Expansion
- Liberal fluid intake to reverse catecholamine-induced volume depletion
Surgical Treatment: Definitive Therapy
Adrenalectomy
- Laparoscopic adrenalectomy: Standard for localized tumors
- Open surgery: Preferred in large or invasive tumors
Intraoperative monitoring of blood pressure and catecholamine levels is essential.
Malignant and Metastatic Pheochromocytoma
Approximately 10% of pheochromocytomas are malignant, diagnosed by metastasis to non-chromaffin tissues (e.g., liver, lungs, bones, lymph nodes).
Treatment Options for Malignancy
- Surgical debulking
- Radiopharmaceutical therapy (I-131 MIBG)
- Chemotherapy: Cyclophosphamide, Vincristine, Dacarbazine (CVD regimen)
- Targeted therapy: Sunitinib, especially in SDHB mutation-positive cases
Prognosis and Follow-Up
With early diagnosis and complete resection, pheochromocytomas have an excellent prognosis. However, recurrence may occur in:
- Genetic cases
- Incomplete tumor removal
- Malignant variants
Lifelong Monitoring
- Annual plasma or urine metanephrine measurements
- Periodic imaging in hereditary cases
- Genetic counseling for patients and first-degree relatives
Prevention and Genetic Counseling
In individuals with hereditary syndromes, prophylactic screening and genetic counseling are crucial. Early detection through family screening enables curative treatment before the onset of symptoms.
Frequently Asked Questions:
Can pheochromocytoma be fatal?
Yes. If undiagnosed or poorly managed, it can lead to life-threatening hypertensive crises or cardiac complications.
Is pheochromocytoma always located in the adrenal gland?
No. Tumors found outside the adrenal glands are called paragangliomas, which share similar features.
Are all pheochromocytomas malignant?
No. Only about 10% of pheochromocytomas are malignant, but malignancy can only be confirmed through evidence of metastasis.
Can pheochromocytoma recur after surgery?
Yes. Recurrence can occur, particularly in hereditary forms, hence the need for lifelong monitoring.
What foods should be avoided with pheochromocytoma?
Foods rich in tyramine (e.g., aged cheese, red wine) may trigger hypertensive episodes and should be limited before diagnosis and surgery.
Pheochromocytoma, though rare, demands a high index of clinical suspicion due to its potentially lethal cardiovascular effects. Through early biochemical testing, precise imaging, and timely surgical intervention, the risk of mortality can be significantly reduced. Long-term follow-up, especially in genetically predisposed individuals, ensures sustained health and mitigates the risk of recurrence or malignancy.