Neurofibromatosis type 1 with plexiform neurofibromas: Neurofibromatosis Type 1 (NF1) is a genetic disorder characterized by the development of multiple nerve sheath tumors, known as neurofibromas. A subset of these, called plexiform neurofibromas (PNs), are more extensive, involve multiple nerve branches, and can cause significant disfigurement, pain, and neurological complications. NF1 is caused by mutations in the NF1 gene, leading to dysregulated cell growth and tumor formation. This article explores the causes, clinical features, diagnostic approaches, and treatment options for NF1 with plexiform neurofibromas.
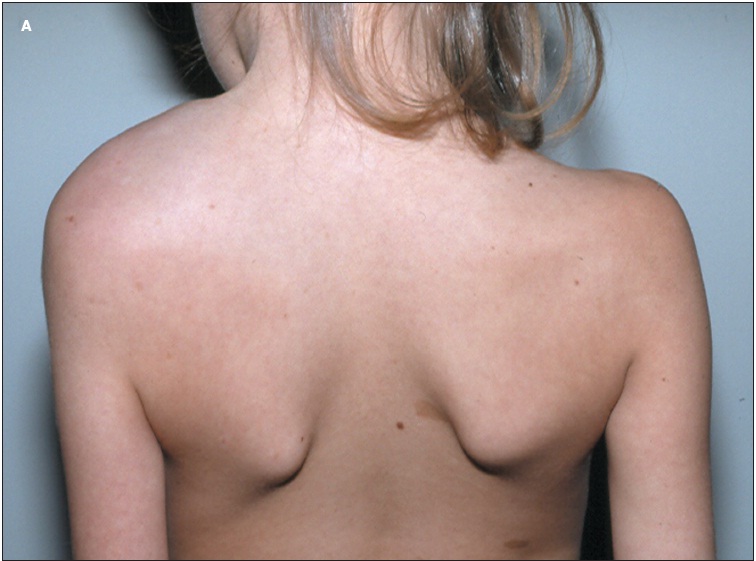
Understanding Neurofibromatosis type 1 with plexiform neurofibromas
1. Genetic Basis and Pathophysiology
NF1 is an autosomal dominant disorder caused by mutations in the NF1 gene, located on chromosome 17q11.2. The NF1 gene encodes neurofibromin, a tumor suppressor protein that regulates cell growth by inhibiting the RAS/MAPK pathway.
- Loss of neurofibromin function leads to uncontrolled proliferation of Schwann cells, fibroblasts, and mast cells, forming benign neurofibromas.
- Plexiform neurofibromas are congenital tumors that arise along multiple nerve branches, infiltrating surrounding tissues.
Clinical Presentation of NF1 with Plexiform Neurofibromas
1. General Symptoms of NF1
NF1 manifests with cutaneous, neurological, and skeletal abnormalities:
- Café-au-lait macules (≥6 spots, >5mm in children, >15mm in adults).
- Freckling in the axillary or inguinal regions.
- Lisch nodules (iris hamartomas).
- Optic gliomas causing vision impairment.
2. Features of Plexiform Neurofibromas (PNs)
PNs are complex, deep-seated nerve sheath tumors that may be:
- Congenital and slow-growing, present from birth but expanding over time.
- Diffuse and invasive, spreading along multiple nerve branches.
- Painful, disfiguring, and functionally impairing, depending on their location.
3. Complications Associated with PNs
- Malignant transformation: ~10-15% of PNs progress to malignant peripheral nerve sheath tumors (MPNSTs).
- Neurological deficits: Compression of nerves can lead to muscle weakness, sensory loss, and mobility issues.
- Orthopedic problems: Scoliosis, limb-length discrepancies, and bone dysplasia.
- Airway obstruction: PNs affecting the head and neck may cause breathing difficulties.
Diagnosis of NF1 with Plexiform Neurofibromas
1. Clinical Criteria for NF1 Diagnosis
According to the National Institutes of Health (NIH) Consensus Criteria, a diagnosis of NF1 requires two or more of the following: Six or more café-au-lait spots Two or more neurofibromas (or one plexiform neurofibroma) Axillary/inguinal freckling Optic glioma Two or more Lisch nodules A first-degree relative with NF1
Six or more café-au-lait spots Two or more neurofibromas (or one plexiform neurofibroma) Axillary/inguinal freckling Optic glioma Two or more Lisch nodules A first-degree relative with NF1
2. Imaging and Biopsy for PNs
- MRI with contrast: Preferred for detecting PNs, their extent, and compression effects.
- PET-CT scan: Used to assess malignant transformation.
- Biopsy: Performed when MPNST is suspected due to rapid tumor growth, pain, or necrosis.
Treatment Strategies for NF1-Associated Plexiform Neurofibromas
1. Medical Management
- MEK inhibitors (Selumetinib): FDA-approved for inoperable plexiform neurofibromas in children. MEK inhibitors target the RAS/MAPK pathway to slow tumor growth.
- Pain management: NSAIDs, opioids, or gabapentinoids for nerve-related pain.
- Steroids: Used for acute nerve compression symptoms.
2. Surgical Intervention
- Indications for Surgery:
- Rapidly growing or painful tumors
- Neurological deficits (e.g., muscle weakness, paralysis)
- Organ compression (e.g., airway obstruction)
- Challenges of PN Resection:
- High risk of nerve damage due to tumor infiltration.
- Incomplete removal often leads to recurrence.
3. Emerging Therapies and Research
- Gene therapy approaches targeting the NF1 gene mutation.
- Immunotherapy trials investigating checkpoint inhibitors for malignant progression.
- Targeted small-molecule inhibitors beyond MEK inhibitors (e.g., PI3K and mTOR inhibitors).
Prognosis and Long-Term Outlook
1. Disease Progression and Quality of Life
- PN growth is unpredictable, and some patients remain asymptomatic.
- Disfigurement and disability impact psychosocial well-being.
- Lifelong monitoring is required due to the risk of malignant transformation.
2. Risk Factors for Malignant Peripheral Nerve Sheath Tumors (MPNSTs)
| Risk Factor | Malignant Potential |
|---|---|
| Large plexiform neurofibromas | High |
| Rapid tumor growth | High |
| Pain and neurological deficits | High |
| Loss of NF1 heterozygosity | High |
Preventive Strategies and Early Intervention
1. Genetic Counseling and Family Screening
- 50% of NF1 cases arise from new mutations, but familial cases require screening.
- Prenatal genetic testing can identify NF1 mutations.
2. Regular Follow-Ups and Surveillance
- Annual neurological and dermatological exams.
- MRI monitoring for growing PNs.
- Early intervention for functional impairments.
Neurofibromatosis Type 1 with plexiform neurofibromas is a complex genetic disorder requiring multidisciplinary management. While surgical and pharmacological options help control symptoms, MEK inhibitors and emerging targeted therapies offer new hope for reducing tumor burden. Ongoing research aims to improve early detection, treatment outcomes, and quality of life for individuals with NF1.