Nephropathic cystinosis is a rare autosomal recessive lysosomal storage disorder characterized by the abnormal accumulation of the amino acid cystine within cells. This accumulation is caused by mutations in the CTNS gene, which encodes the lysosomal cystine transporter, cystinosin. The disease primarily affects the kidneys but can extend to multiple organ systems, leading to severe complications if left untreated.
Genetic and Molecular Basis of Nephropathic Cystinosis
Mutation in the CTNS Gene
Nephropathic cystinosis is inherited in an autosomal recessive manner. Both parents must be carriers of a defective CTNS gene for a child to inherit the disease. The CTNS gene, located on chromosome 17p13, encodes cystinosin, which is responsible for exporting cystine out of lysosomes. Mutations impair this function, causing cystine to accumulate and form crystals that damage cellular structures.
Pathophysiology
Cystine buildup in lysosomes triggers cellular dysfunction, particularly in renal proximal tubules, leading to renal Fanconi syndrome. Over time, this pathology extends to muscles, eyes, thyroid, pancreas, and the central nervous system.
Clinical Manifestations
Early Onset Symptoms
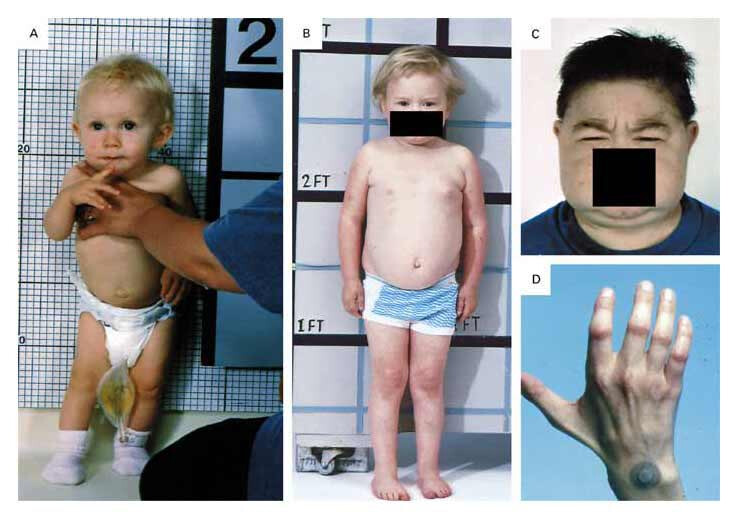
Nephropathic cystinosis typically manifests in infancy, between 6 and 12 months of age. Initial symptoms include:
- Polyuria and polydipsia
- Dehydration
- Failure to thrive
- Rickets due to phosphate loss
- Vomiting and constipation
Progressive Multisystem Involvement
If left untreated, the disease advances with systemic manifestations, including:
- Photophobia due to corneal cystine crystals
- Hypothyroidism
- Myopathy and muscle wasting
- Diabetes mellitus
- Swallowing difficulties (dysphagia)
- Neurological impairment
Renal Involvement and Fanconi Syndrome
Renal Tubular Dysfunction
Renal Fanconi syndrome is a hallmark of nephropathic cystinosis and is characterized by the loss of essential nutrients through the urine, including:
- Glucose (glycosuria)
- Phosphate (hypophosphatemia)
- Bicarbonate (metabolic acidosis)
- Amino acids (aminoaciduria)
- Calcium and potassium
Chronic Kidney Disease Progression
Without intervention, chronic kidney disease (CKD) progresses rapidly. Most untreated children require renal replacement therapy or kidney transplantation by the end of the first decade of life.
Diagnostic Strategies
Clinical Evaluation
Diagnosis begins with a thorough clinical history and physical examination focusing on early signs such as poor growth and electrolyte imbalances.
Laboratory Tests
- Elevated cystine levels in white blood cells (WBC cystine assay)
- Urinary loss of solutes
- Blood tests showing electrolyte abnormalities
Genetic Testing
Molecular analysis confirms diagnosis by identifying mutations in the CTNS gene. Carrier screening and prenatal diagnosis are available for at-risk families.
Imaging and Ophthalmologic Exams
- Slit-lamp examination reveals pathognomonic corneal cystine crystals.
- Renal ultrasound may show nephrocalcinosis and kidney shrinkage.
Therapeutic Approaches
Cysteamine Therapy
The cornerstone of treatment is oral cysteamine bitartrate, which depletes cystine from lysosomes by converting it into cysteine-cysteamine mixed disulfide that can exit via existing transporters.
- Dosing: Administered every 6 hours to maintain optimal cystine depletion.
- Monitoring: Regular WBC cystine level checks to ensure efficacy.
Topical cysteamine eye drops are used to dissolve corneal crystals and improve photophobia.
Supportive Management
- Electrolyte replacement (phosphate, potassium, bicarbonate)
- Vitamin D and calcium supplements for rickets
- Growth hormone therapy in growth-retarded children
- Levothyroxine for hypothyroidism
- Insulin for diabetes mellitus
- Speech and physical therapy for neuromuscular complications
Renal Transplantation
In cases of end-stage renal disease, kidney transplantation is effective. Post-transplant cysteamine therapy must continue to prevent extra-renal complications.
Long-Term Prognosis
Prognosis has improved significantly with early diagnosis and consistent cysteamine therapy. Many patients now live into adulthood with preserved organ function. However, lifelong treatment adherence is critical to delay progression and maintain quality of life.
Research and Emerging Therapies
Recent studies are exploring gene therapy, improved drug formulations, and stem cell approaches. Ongoing clinical trials aim to reduce dosing frequency and enhance systemic delivery of cysteamine or develop mutation-specific treatments.
Living with Nephropathic Cystinosis
Psychosocial Impact
Patients and caregivers face substantial challenges including medical adherence, developmental delays, and financial burdens. Multidisciplinary care involving nephrologists, geneticists, endocrinologists, and psychologists is essential.
Patient Advocacy and Support
Organizations like the Cystinosis Research Network and Global Genes offer educational resources, funding, and community support for families managing cystinosis.
Frequently Asked Questions:
What is nephropathic cystinosis?
Nephropathic cystinosis is a genetic lysosomal storage disorder that leads to cystine accumulation in cells, causing kidney and systemic damage.
How is cystinosis diagnosed?
Diagnosis is based on clinical symptoms, elevated WBC cystine levels, slit-lamp examination for corneal crystals, and genetic testing of the CTNS gene.
Can cystinosis be cured?
There is no cure, but cysteamine therapy can manage the condition effectively and prevent complications when started early.
What organs are affected by nephropathic cystinosis?
The kidneys, eyes, thyroid, pancreas, muscles, and brain can all be affected as the disease progresses.
Is cystinosis fatal?
With early diagnosis and continuous treatment, many patients live into adulthood with improved quality of life and delayed disease progression.
Nephropathic cystinosis, while rare and complex, is a manageable condition with early diagnosis and rigorous treatment. Advances in therapeutics and growing awareness are transforming outcomes for patients globally. Continued research and support will be vital to further improve prognosis and quality of life.