Neoplasms Associated with von Hippel Lindau Disease: Von Hippel-Lindau (VHL) disease is a rare, autosomal dominant hereditary cancer syndrome characterized by the development of benign and malignant tumors in multiple organ systems. This multisystem disorder arises due to germline mutations in the VHL tumor suppressor gene located on chromosome 3p25.3, which plays a critical role in the regulation of hypoxia-inducible factors (HIFs) and angiogenesis. The inactivation of the VHL gene leads to uncontrolled cell proliferation, angiogenesis, and tumor formation.
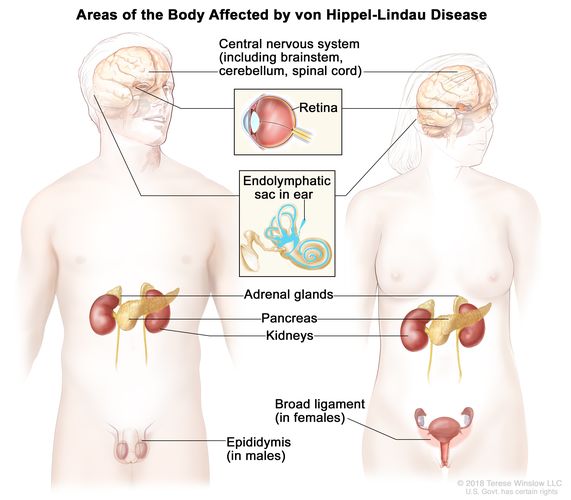
Major Neoplasms Associated with VHL Disease
Central Nervous System Hemangioblastomas
CNS hemangioblastomas are the most common tumors found in patients with VHL. These highly vascular tumors primarily affect the cerebellum, brainstem, and spinal cord, often presenting between the second and fourth decades of life. Symptoms depend on tumor location and may include ataxia, headaches, and spinal cord compression.
MRI remains the gold standard for diagnosis, revealing cystic lesions with enhancing mural nodules. Surgical resection is often curative for symptomatic tumors.
Retinal Hemangioblastomas
Retinal hemangioblastomas, often the first manifestation of VHL disease, are capillary hemangiomas affecting the peripheral retina or optic nerve head. If left untreated, they can cause vision loss due to exudative retinal detachment or macular edema.
Early detection through annual ophthalmic evaluations and treatment via laser photocoagulation or cryotherapy is essential to preserve visual function.
Renal Cell Carcinoma (Clear Cell Type)
Renal cell carcinoma (RCC), particularly the clear cell subtype, is a life-threatening malignancy seen in up to 70% of VHL patients. RCCs typically arise from pre-existing renal cysts and may be multifocal and bilateral.
Regular abdominal imaging (ultrasound, CT, or MRI) is critical for early detection. Nephron-sparing surgeries are preferred to preserve renal function while controlling tumor burden.
Pancreatic Neuroendocrine Tumors
Pancreatic neuroendocrine tumors (pNETs) occur in approximately 10–20% of individuals with VHL. These tumors are generally non-functioning and asymptomatic, though some may secrete hormones, leading to clinical syndromes.
Management involves surveillance for tumors <3 cm and surgical resection for larger or symptomatic masses. Fine-needle aspiration and chromogranin A levels aid in diagnosis.
Pheochromocytomas and Paragangliomas
Pheochromocytomas, found in the adrenal medulla, and extra-adrenal paragangliomas are significant features of VHL disease. These tumors may secrete catecholamines, causing hypertension, tachycardia, and headaches.
Biochemical screening (plasma free metanephrines) and imaging (MRI or MIBG scintigraphy) confirm the diagnosis. Surgical resection is the treatment of choice, often preceded by alpha-adrenergic blockade.
Endolymphatic Sac Tumors
Endolymphatic sac tumors (ELSTs) are low-grade adenocarcinomas of the inner ear associated with sensorineural hearing loss, tinnitus, and vertigo. They can cause progressive hearing impairment if not addressed.
High-resolution CT and MRI are essential for diagnosis. Surgical resection may be required to prevent further auditory damage.
Epididymal and Broad Ligament Cystadenomas
In males, epididymal cystadenomas are benign tumors of the epididymis, often bilateral. In females, similar cystadenomas may develop in the broad ligament. These tumors are generally asymptomatic but can cause discomfort or infertility.
Ultrasound imaging helps in identification, with surgery reserved for symptomatic cases.
Pathogenesis: The Role of VHL Gene and HIF Pathway
The VHL gene encodes pVHL, a protein critical for the degradation of hypoxia-inducible factor alpha (HIF-α). In normoxia, pVHL binds to HIF-α, targeting it for ubiquitination and proteasomal degradation. Loss of pVHL function leads to accumulation of HIF-α, promoting angiogenesis and tumorigenesis.
Genetic Testing and Surveillance
Early genetic diagnosis is critical in individuals with a family history of VHL. Once a VHL gene mutation is confirmed, comprehensive surveillance must begin in childhood to detect neoplasms early.
Recommended Surveillance Includes:
- Annual ophthalmologic exams from infancy
- MRI of the CNS every 2 years starting from adolescence
- Abdominal imaging (ultrasound or MRI) annually
- Annual plasma metanephrine screening for pheochromocytomas
Treatment and Management Strategies
Multidisciplinary Approach
Management of VHL neoplasms necessitates a multidisciplinary approach, involving neurologists, nephrologists, oncologists, ophthalmologists, and genetic counselors. The treatment strategy varies depending on tumor type, size, location, and symptomatology.
- Surgical excision is standard for CNS tumors, RCCs, and pheochromocytomas.
- Minimally invasive techniques such as radiofrequency ablation or cryoablation are options for small renal tumors.
- Targeted therapies, like VEGF inhibitors (e.g., belzutifan), have shown efficacy in managing multiple tumor types in VHL.
Prognosis and Life Expectancy
With early detection and timely intervention, individuals with VHL disease can maintain a near-normal life expectancy. However, mortality is most often due to complications from CNS hemangioblastomas or metastatic RCC.
Long-term prognosis is significantly improved through adherence to surveillance protocols and individualized treatment plans.
Von Hippel-Lindau disease presents a complex clinical picture marked by the formation of neoplasms in multiple organ systems. Understanding the molecular basis, implementing early surveillance, and applying targeted treatment modalities are critical to improving outcomes in affected individuals. With advancements in genetic testing and targeted therapies, the prognosis for VHL patients continues to improve, reinforcing the importance of precision medicine in hereditary cancer syndromes.