Mucopolysaccharidosis type VII (MPS VII), also known as Sly syndrome, is a rare lysosomal storage disorder caused by a deficiency of the β-glucuronidase (GUSB) enzyme. This enzyme is responsible for breaking down glycosaminoglycans (GAGs) such as dermatan sulfate, heparan sulfate, and chondroitin sulfate. The accumulation of these substances leads to progressive multisystem complications, affecting the skeletal system, organs, and cognitive function.
What is Mucopolysaccharidosis Type VII?
MPS VII is part of the mucopolysaccharidosis family, a group of inherited disorders caused by enzyme deficiencies in the lysosomal degradation of glycosaminoglycans. It follows an autosomal recessive inheritance pattern, meaning an affected individual inherits two defective copies of the GUSB gene, one from each parent.
Genetic Basis of MPS VII
MPS VII results from mutations in the GUSB gene, located on chromosome 7q11.21. These mutations lead to a deficiency or complete absence of β-glucuronidase, causing the accumulation of GAGs in tissues and organs, ultimately leading to progressive damage.
Symptoms of MPS VII
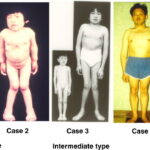
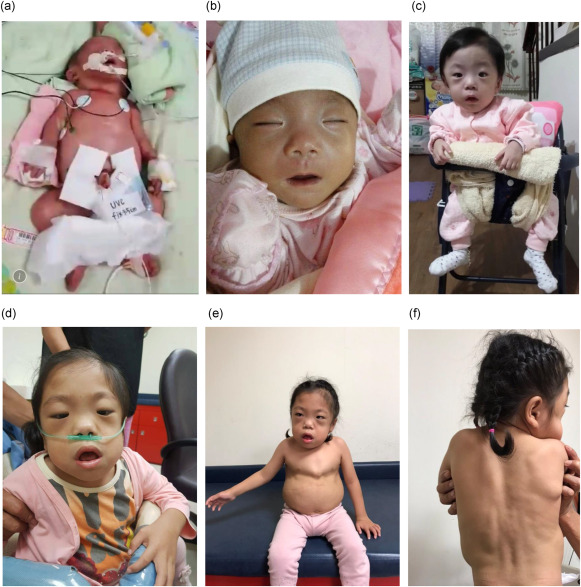
MPS VII has a wide spectrum of severity, ranging from hydrops fetalis (severe prenatal form) to mild adult-onset cases. Symptoms typically appear in early childhood and worsen over time.
Common Symptoms
- Coarse facial features (thickened lips, broad nose, enlarged tongue)
- Short stature and skeletal abnormalities
- Joint stiffness and restricted movement
- Hepatosplenomegaly (enlarged liver and spleen)
- Progressive vision and hearing loss
- Cognitive impairment (varies by severity)
- Frequent respiratory infections
Skeletal and Musculoskeletal Complications
- Dysostosis multiplex (skeletal dysplasia seen in MPS disorders)
- Kyphoscoliosis (curved spine)
- Genu valgum (knock-knees)
- Joint contractures leading to mobility issues
Neurological and Cognitive Impact
- Hydrocephalus (fluid buildup in the brain)
- Developmental delays
- Variable cognitive impairment
Respiratory and Cardiovascular Issues
- Airway obstruction due to thickened tissues
- Sleep apnea
- Cardiac abnormalities (valvular disease, cardiomyopathy)
Diagnosis of MPS VII
Early diagnosis is essential for effective management.
Clinical Examination
- Growth abnormalities
- Skeletal deformities
- Hepatosplenomegaly detection
Laboratory Tests
- Enzyme Activity Assay: Measures β-glucuronidase enzyme levels in blood or fibroblasts.
- Urinary GAG Analysis: Detects excessive dermatan sulfate, heparan sulfate, and chondroitin sulfate.
- Genetic Testing: Confirms GUSB gene mutations.
Imaging and Additional Testing
- X-rays: Show characteristic dysostosis multiplex.
- MRI: Identifies hydrocephalus or spinal cord compression.
- Echocardiogram: Evaluates heart valve abnormalities.
Treatment Options for MPS VII
There is no cure for MPS VII, but treatments focus on symptom management and improving quality of life.
1. Enzyme Replacement Therapy (ERT)
Vestronidase alfa (MEPSEVII) is the first FDA-approved ERT for MPS VII.
- Administered via intravenous infusion every two weeks.
- Reduces GAG accumulation and improves mobility.
- Does not cross the blood-brain barrier, so it does not address cognitive symptoms.
2. Supportive and Symptomatic Treatments
- Physical Therapy: Helps maintain joint mobility and reduce stiffness.
- Respiratory Support: CPAP, BiPAP, or tracheostomy for airway management.
- Hearing Aids: Address progressive hearing loss.
- Pain Management: Medications and physical therapy for joint pain.
3. Surgical Interventions
- Spinal Decompression Surgery: Prevents spinal cord compression.
- Corneal Transplant: Improves vision in severe cases of corneal clouding.
- Cardiac Surgery: Valve replacement if severe heart disease develops.
Prognosis and Life Expectancy
MPS VII prognosis depends on disease severity.
- Severe cases (hydrops fetalis): Often fatal in infancy.
- Moderate cases: Survival into adolescence or early adulthood with supportive care.
- Milder cases: Individuals may live into adulthood with medical management.
Research and Emerging Therapies
Ongoing studies aim to develop more effective treatments:
- Gene Therapy: Investigating ways to deliver functional GUSB genes to patient cells.
- Substrate Reduction Therapy (SRT): Targeting GAG synthesis reduction.
- Chaperone Therapy: Exploring drugs to stabilize β-glucuronidase enzyme activity.
Mucopolysaccharidosis type VII (Sly syndrome) is a rare and progressive genetic disorder caused by β-glucuronidase deficiency. While enzyme replacement therapy and supportive treatments can manage symptoms, ongoing research is crucial for improving outcomes. Early diagnosis and multidisciplinary care are essential to enhancing quality of life.