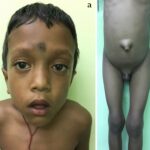
Mucopolysaccharidosis type I (MPS I) is a rare genetic disorder caused by a deficiency of the enzyme alpha-L-iduronidase. This deficiency leads to the accumulation of glycosaminoglycans (GAGs) in various tissues, resulting in progressive organ damage. MPS I encompasses a spectrum of severity, historically classified into Hurler syndrome (severe), Hurler-Scheie syndrome (intermediate), and Scheie syndrome (mild). Understanding the underlying causes, symptoms, and available treatments is crucial for improving patient outcomes.
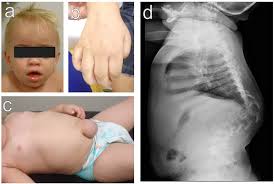
What is Mucopolysaccharidosis Type I (MPS I)?
MPS I is a lysosomal storage disorder that results from mutations in the IDUA gene, which encodes the enzyme alpha-L-iduronidase. This enzyme is responsible for breaking down dermatan sulfate and heparan sulfate, two types of glycosaminoglycans. Without functional alpha-L-iduronidase, these substances accumulate in cells, leading to progressive tissue and organ dysfunction.
Classification of MPS I
| Type | Severity | Symptoms | Life Expectancy |
|---|---|---|---|
| Hurler Syndrome (Severe MPS I) | Most severe | Severe developmental delays, skeletal abnormalities, organ enlargement | Often fatal in early childhood |
| Hurler-Scheie Syndrome (Intermediate MPS I) | Moderate | Milder intellectual disability, joint stiffness, corneal clouding | May survive into adulthood |
| Scheie Syndrome (Mild MPS I) | Least severe | Normal intelligence, joint stiffness, cardiac issues | Near-normal lifespan |
Causes and Genetic Basis of MPS I
MPS I is an autosomal recessive disorder, meaning an individual must inherit two copies of the defective IDUA gene (one from each parent) to develop the condition. Carriers, who have only one defective copy, do not exhibit symptoms.
Symptoms of MPS I
MPS I affects multiple organ systems, with symptoms worsening over time. The severity depends on whether the individual has Hurler, Hurler-Scheie, or Scheie syndrome.
Early Symptoms
- Developmental delays
- Frequent respiratory infections
- Enlarged liver and spleen (hepatosplenomegaly)
- Skeletal abnormalities (dysostosis multiplex)
- Coarse facial features
Neurological Symptoms
- Progressive intellectual disability (more severe in Hurler syndrome)
- Hydrocephalus (fluid accumulation in the brain)
- Carpal tunnel syndrome due to nerve compression
Musculoskeletal Symptoms
- Joint stiffness and limited mobility
- Spinal abnormalities such as kyphosis
- Short stature
Cardiovascular and Respiratory Issues
- Heart valve abnormalities
- Airway obstruction due to enlarged tonsils and adenoids
- Sleep apnea
Ophthalmological Symptoms
- Corneal clouding, leading to vision impairment
- Retinal degeneration in severe cases
Diagnosis of MPS I
Clinical Examination and Family History
A physician may suspect MPS I based on a combination of physical features and developmental delays.
Laboratory and Genetic Tests
- Enzyme Activity Test: Measures alpha-L-iduronidase levels in blood or fibroblasts.
- Urinary Glycosaminoglycan (GAG) Analysis: Detects elevated dermatan and heparan sulfate.
- Genetic Testing: Confirms mutations in the IDUA gene to establish a definitive diagnosis.
Imaging and Other Tests
- X-rays: Identify skeletal abnormalities.
- Echocardiogram: Assesses heart function.
- MRI/CT Scan: Evaluates brain and spinal cord involvement.
Treatment Options for MPS I
While there is no cure for MPS I, treatment focuses on managing symptoms, slowing disease progression, and improving quality of life.
1. Enzyme Replacement Therapy (ERT)
ERT with laronidase (Aldurazyme) provides the missing enzyme to help break down glycosaminoglycans.
- Administered via intravenous infusion.
- Reduces organ enlargement and improves joint mobility.
- Does not significantly cross the blood-brain barrier, limiting neurological benefits.
2. Hematopoietic Stem Cell Transplantation (HSCT)
- Recommended for severe cases (Hurler syndrome) early in life.
- Can slow or prevent neurological decline if performed before significant brain damage occurs.
- High-risk procedure requiring careful evaluation.
3. Supportive Treatments
- Physical Therapy: Maintains joint mobility and reduces stiffness.
- Surgical Interventions: Corrects airway obstructions, hernias, and heart valve abnormalities.
- Hearing Aids & Vision Support: Addresses sensory impairments.
- Respiratory Support: CPAP or tracheostomy for severe airway obstruction.
Prognosis and Life Expectancy
The prognosis for MPS I varies depending on the severity of the disease:
- Hurler Syndrome: Without treatment, life expectancy is often less than 10 years. With early HSCT, survival improves significantly.
- Hurler-Scheie Syndrome: Can live into adulthood with supportive care.
- Scheie Syndrome: Normal or near-normal lifespan, though complications like cardiac issues may arise.
Research and Emerging Therapies
Ongoing research aims to improve treatment options:
- Gene Therapy: Potential for a long-term cure by delivering a functional IDUA gene.
- Small Molecule Therapy: Investigating drugs that can cross the blood-brain barrier to address neurological symptoms.
Mucopolysaccharidosis type I is a complex genetic disorder with a wide spectrum of severity. Early diagnosis and intervention, particularly through enzyme replacement therapy and stem cell transplantation, can significantly improve outcomes. Continued research offers hope for more effective treatments in the future, enhancing the quality of life for affected individuals.