Lysosomal alpha-1,4-glucosidase deficiency, also known as Pompe disease or Glycogen Storage Disease Type II (GSD II), is a rare genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen into glucose within lysosomes. Without sufficient GAA, glycogen accumulates in muscle tissues, leading to progressive muscle weakness and organ dysfunction.
Causes and Genetic Basis of Pompe Disease
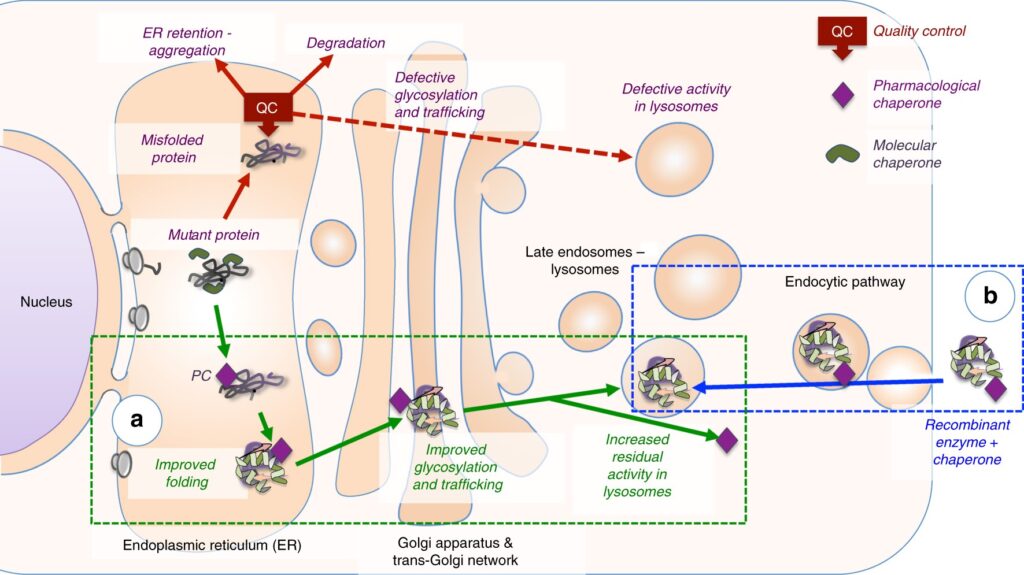
Pompe disease is caused by mutations in the GAA gene, which encodes the lysosomal enzyme acid alpha-glucosidase. This disorder is inherited in an autosomal recessive pattern, meaning both parents must carry and pass on a defective gene for the child to develop the disease.
Types of Pompe Disease
Pompe disease is categorized into three main types based on the severity and age of onset:
- Classic Infantile-Onset Pompe Disease (IOPD): Appears within the first few months of life, characterized by severe muscle weakness, heart enlargement, and respiratory failure.
- Non-Classic Infantile-Onset Pompe Disease: Manifests in late infancy with delayed motor development and progressive muscle weakness.
- Late-Onset Pompe Disease (LOPD): Symptoms appear during childhood or adulthood, primarily affecting skeletal and respiratory muscles.
Symptoms of Lysosomal Alpha-1,4-Glucosidase Deficiency
The severity of Pompe disease symptoms depends on the age of onset and the level of residual enzyme activity.
Symptoms of Infantile-Onset Pompe Disease
- Severe hypotonia (muscle weakness)
- Cardiomegaly (enlarged heart)
- Respiratory distress
- Feeding difficulties and failure to thrive
- Delayed motor milestones
Symptoms of Late-Onset Pompe Disease
- Progressive muscle weakness, particularly in the legs and trunk
- Respiratory insufficiency and shortness of breath
- Difficulty walking or climbing stairs
- Fatigue and exercise intolerance
- Scoliosis and joint contractures in severe cases
Diagnosis of Pompe Disease
Diagnosing Pompe disease involves multiple laboratory, imaging, and genetic tests.
Biochemical Tests
- Acid Alpha-Glucosidase Enzyme Assay: Measures enzyme activity in blood or fibroblasts.
- Blood Biomarkers: Elevated levels of creatine kinase (CK), lactate dehydrogenase (LDH), and aspartate aminotransferase (AST) may indicate muscle damage.
Imaging and Functional Tests
- Electromyography (EMG): Identifies myopathic changes in muscle function.
- MRI of Skeletal Muscles: Detects patterns of muscle degeneration and fatty infiltration.
Genetic Testing
- GAA Gene Sequencing: Confirms mutations responsible for Pompe disease.
- Carrier Testing: Recommended for family members of affected individuals.
Treatment and Management Strategies
While Pompe disease has no cure, enzyme replacement therapy (ERT) and supportive care significantly improve disease outcomes.
Enzyme Replacement Therapy (ERT)
- Alglucosidase Alfa (Myozyme®/Lumizyme®): A recombinant human GAA enzyme that reduces glycogen accumulation.
- Administered through intravenous infusions every two weeks.
- Improves muscle function, respiratory health, and overall survival in infantile-onset cases.
Supportive Treatments
- Physical Therapy: Strengthens muscles and maintains mobility.
- Respiratory Support: Non-invasive ventilation (NIV) or mechanical ventilation for individuals with respiratory failure.
- Nutritional Management: High-protein diets and enteral feeding for those with swallowing difficulties.
- Orthopedic Interventions: Corrective surgeries for scoliosis or joint contractures.
Prognosis and Life Expectancy
- Infantile-Onset Pompe Disease: Without treatment, most infants do not survive beyond one year due to respiratory and cardiac complications.
- Late-Onset Pompe Disease: Progression varies, but early intervention with ERT and supportive care can significantly improve quality of life.
Genetic Counseling and Preventive Measures
- Prenatal Testing: Chorionic villus sampling (CVS) or amniocentesis for high-risk pregnancies.
- Newborn Screening: Implemented in several countries for early detection and timely treatment.
- Carrier Screening: Recommended for families with a history of Pompe disease.
Lysosomal Alpha-1,4-Glucosidase Deficiency, or Pompe disease, is a debilitating genetic disorder requiring early diagnosis and prompt medical intervention. Advances in enzyme replacement therapy (ERT) have transformed disease outcomes, extending survival and enhancing quality of life. Genetic screening, early detection, and multidisciplinary care remain crucial in managing this complex condition.