Lysosomal Acid Lipase Deficiency (LAL-D) is a rare genetic disorder affecting the body’s ability to break down lipids. The condition results from mutations in the LIPA gene, which encodes the enzyme lysosomal acid lipase (LAL). This enzyme is crucial for breaking down cholesterol esters and triglycerides within lysosomes.
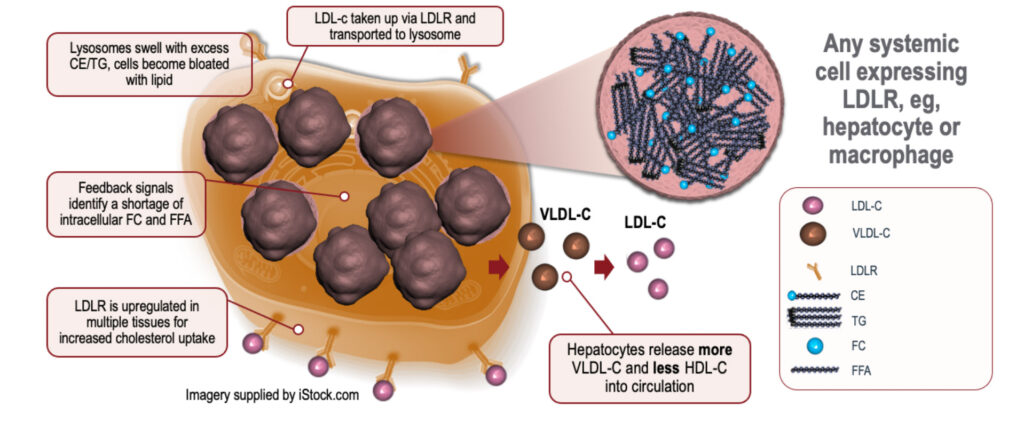
Causes and Genetic Basis of LAL-D
LAL-D is caused by mutations in the LIPA gene, leading to deficient or non-functional lysosomal acid lipase enzyme. Without this enzyme, lipids accumulate in various organs, particularly the liver, spleen, and blood vessels. LAL-D is inherited in an autosomal recessive pattern, meaning an affected individual must inherit two faulty copies of the gene, one from each parent.
Types of LAL-D
LAL-D presents in two primary forms:
- Wolman Disease (Early-Onset LAL-D): Appears in infancy, characterized by severe fat accumulation, liver dysfunction, and rapid disease progression.
- Cholesteryl Ester Storage Disease (CESD) (Late-Onset LAL-D): Milder form, often diagnosed in children or adults, leading to progressive liver damage and cardiovascular complications.
Symptoms of Lysosomal Acid Lipase Deficiency
The symptoms of LAL-D vary depending on the severity of the enzyme deficiency. Common symptoms include:
Symptoms in Infants (Wolman Disease)
- Severe hepatosplenomegaly (enlarged liver and spleen)
- Persistent vomiting and diarrhea
- Failure to thrive
- Severe malabsorption
- Calcification of adrenal glands
- Rapid disease progression leading to fatal outcomes
Symptoms in Children and Adults (CESD)
- Hepatomegaly (enlarged liver)
- Increased liver enzymes (ALT and AST)
- Elevated cholesterol and LDL levels
- Fatty liver disease (NAFLD/NASH)
- Progressive liver fibrosis or cirrhosis
- Early-onset atherosclerosis
Diagnosis of LAL-D
Diagnosing LAL-D involves multiple laboratory and genetic tests:
Biochemical Tests
- LAL Enzyme Activity Test: Measures the functional activity of the lysosomal acid lipase enzyme in blood.
- Liver Function Tests (LFTs): Evaluates ALT, AST, and bilirubin levels.
- Lipid Panel: Assesses cholesterol levels, especially elevated LDL and reduced HDL.
Imaging and Histopathology
- Liver Biopsy: Reveals lipid accumulation and fibrosis.
- Abdominal Ultrasound/CT Scan: Detects hepatosplenomegaly and fatty liver.
Genetic Testing
- LIPA Gene Sequencing: Confirms pathogenic mutations responsible for LAL-D.
Treatment and Management Strategies
LAL-D has no complete cure, but enzyme replacement therapy (ERT) significantly improves disease outcomes.
Enzyme Replacement Therapy (ERT)
- Sebelipase Alfa (Kanuma™): A recombinant human LAL enzyme that helps restore lipid metabolism.
- Administered intravenously, typically every one to two weeks.
- Improves liver function and reduces lipid accumulation.
Supportive Treatments
- Lipid-Lowering Medications: Statins and fibrates help manage high cholesterol levels.
- Liver Disease Management: Monitoring for cirrhosis, fibrosis, or hepatic complications.
- Nutritional Support: Low-fat diet with controlled cholesterol intake.
Liver Transplantation
In severe cases, particularly for infants with Wolman disease, liver transplantation may be necessary to prolong survival.
Prognosis and Life Expectancy
- Infantile LAL-D (Wolman Disease): Without treatment, most infants do not survive past the first year.
- Childhood/Adult LAL-D (CESD): Prognosis varies; with ERT and lifestyle management, individuals can lead relatively normal lives while managing liver and cardiovascular risks.
Preventive Measures and Genetic Counseling
- Carrier Testing: Recommended for at-risk families with a history of LAL-D.
- Prenatal Testing: Genetic screening through amniocentesis or chorionic villus sampling (CVS) for high-risk pregnancies.
- Early Diagnosis: Initiating enzyme replacement therapy early can prevent severe complications.
Lysosomal Acid Lipase Deficiency (LAL-D) is a life-threatening genetic disorder requiring early detection and appropriate medical intervention. With advancements in enzyme replacement therapy (ERT), disease progression can be significantly slowed, improving survival rates and quality of life. Individuals with a family history should seek genetic counseling and early screening to prevent complications associated with this disorder.