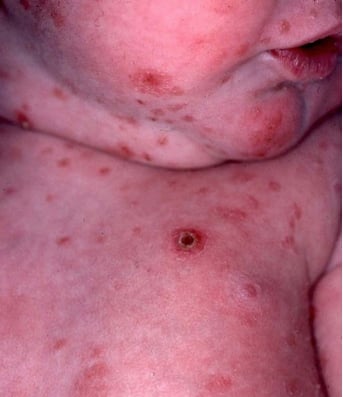
Letterer-Siwe disease, a severe form of Langerhans cell histiocytosis (LCH), predominantly affects children under the age of two. This rare and aggressive disorder is characterized by the rapid proliferation of Langerhans cells, leading to multisystem involvement and a challenging clinical course.
Etiology and Pathogenesis
The precise cause of Letterer-Siwe disease remains under investigation. It is understood to involve the abnormal accumulation and proliferation of Langerhans cells—specialized dendritic cells involved in antigen presentation. These cells infiltrate various tissues, forming granulomatous lesions that disrupt normal organ function. Genetic mutations, particularly in the BRAF and MAP2K1 genes, have been implicated in the pathogenesis, suggesting an oncogenic component to the disease.
Clinical Presentation
Letterer-Siwe disease manifests with a spectrum of symptoms due to its multisystem involvement:
- Dermatological Signs: Early indicators often include scaly, seborrheic, or eczematous rashes, primarily affecting the scalp, ear canals, abdomen, and intertriginous areas.
- Hematologic and Systemic Symptoms: Patients may present with anemia, neutropenia, and thrombocytopenia, leading to fatigue, increased infection susceptibility, and bleeding tendencies.
- Lymphadenopathy and Hepatosplenomegaly: Enlargement of lymph nodes, liver, and spleen is common, contributing to abdominal distension and discomfort.
- Skeletal Involvement: Osteolytic bone lesions can cause pain and increase fracture risk.
- Pulmonary Complications: Respiratory distress may occur due to lung involvement, presenting as cough and tachypnea.
In severe cases, progression can lead to organ failure and sepsis, underscoring the disease’s aggressive nature.
Diagnostic Approach
Diagnosing Letterer-Siwe disease necessitates a comprehensive evaluation:
- Histopathological Examination: Biopsy of affected tissues reveals infiltration by Langerhans cells. Immunohistochemical staining positive for CD1a, CD207 (langerin), and S-100 protein confirms the diagnosis.
- Imaging Studies: X-rays, CT scans, and MRIs assist in identifying osteolytic lesions and assessing the extent of organ involvement.
- Laboratory Tests: Complete blood counts may show cytopenias; liver function tests can indicate hepatic involvement.
Early and accurate diagnosis is critical for initiating appropriate treatment and improving outcomes.
Treatment Strategies
Management of Letterer-Siwe disease is tailored to disease severity and organ involvement:
- Chemotherapy: Systemic chemotherapy remains the cornerstone of treatment, aiming to reduce Langerhans cell proliferation.
- Targeted Therapy: In cases with identified BRAF mutations, targeted inhibitors like vemurafenib have shown promise.
- Supportive Care: Addressing anemia, preventing infections, and managing organ dysfunction are vital components of comprehensive care.
Given the disease’s complexity, a multidisciplinary approach involving pediatric oncologists, dermatologists, and other specialists is essential.
Prognosis
The prognosis for Letterer-Siwe disease varies:
- Age and Disease Extent: Younger patients with extensive organ involvement generally have a poorer prognosis.
- Treatment Response: Early response to therapy is a critical prognostic indicator; patients showing improvement during initial treatment phases tend to have better outcomes.
Despite advances in treatment, the aggressive nature of Letterer-Siwe disease necessitates ongoing research to develop more effective therapies and improve survival rates.
Understanding Letterer-Siwe disease’s complexities is vital for timely diagnosis and intervention. Through continued research and clinical collaboration, we aim to enhance patient outcomes and provide hope for affected families.