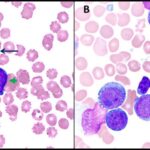
Langerhans Cell Histiocytosis (LCH) is a rare disorder characterized by the abnormal proliferation of Langerhans cells, a type of dendritic cell involved in the immune response. These abnormal cells accumulate in various tissues, leading to damage and dysfunction.
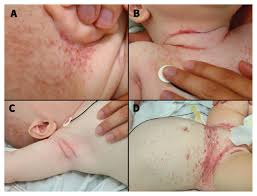
Causes and Risk Factors
The exact cause of LCH remains unclear, but research suggests it may have both inflammatory and neoplastic properties. Genetic mutations, particularly in the BRAF V600E gene, have been linked to the disease. Risk factors include:
- Genetic predisposition
- Environmental triggers
- Viral infections affecting immune regulation
Symptoms of Langerhans Cell Histiocytosis
LCH symptoms vary based on the organs involved. Common manifestations include:
- Bone involvement: Pain, fractures, and skull lesions
- Skin lesions: Rash, scaly patches, and ulcers
- Lung involvement: Chronic cough, difficulty breathing
- Endocrine system: Diabetes insipidus, growth hormone deficiency
- Liver and spleen: Hepatomegaly, jaundice, anemia
- Central nervous system (CNS): Neurodegenerative symptoms, seizures
Types of Langerhans Cell Histiocytosis
LCH can be classified into different forms based on its extent and severity:
- Single-System LCH (SS-LCH): Affects only one organ system (e.g., bone or skin).
- Multisystem LCH (MS-LCH): Involves multiple organ systems with varying severity.
- High-Risk LCH: Affects vital organs such as the liver, spleen, and bone marrow.
Diagnosis of LCH
The diagnosis of LCH involves multiple tests to confirm histiocytic proliferation and genetic mutations. Standard diagnostic methods include:
- Histopathological examination: Biopsy of affected tissue with immunohistochemical staining for CD1a and S100 proteins
- Imaging tests: X-rays, CT scans, MRI, and PET scans to assess organ involvement
- Genetic testing: Detection of BRAF V600E or MAP2K1 mutations
- Bone marrow biopsy: Required in high-risk cases
Treatment Approaches for LCH
Treatment depends on disease severity and organ involvement. Standard treatment options include:
1. Observation and Supportive Care
For mild, self-limiting cases (e.g., skin lesions), close monitoring without immediate treatment may be recommended.
2. Chemotherapy
For multisystem or high-risk cases, chemotherapy is the primary treatment:
- Vinblastine and prednisone (first-line therapy)
- Cytarabine or cladribine (for refractory cases)
- Methotrexate or mercaptopurine (for severe cases)
3. Targeted Therapy
Recent advances have introduced targeted inhibitors:
- BRAF inhibitors (vemurafenib, dabrafenib) for BRAF V600E mutations
- MEK inhibitors (trametinib) for MAP2K1 mutations
4. Radiation Therapy
Low-dose radiation may be considered for localized bone lesions or CNS involvement.
5. Stem Cell Transplantation
For severe, treatment-resistant LCH, hematopoietic stem cell transplantation (HSCT) may be an option.
Prognosis and Long-Term Outlook
The prognosis of LCH depends on disease extent and response to treatment:
- Single-system LCH: Good prognosis with high survival rates
- Multisystem LCH: Variable outcomes depending on organ involvement
- High-risk LCH: Requires aggressive treatment but survival rates have improved with targeted therapies
Langerhans Cell Histiocytosis is a rare yet complex disorder with a spectrum of clinical manifestations. Early diagnosis and targeted therapies have significantly improved patient outcomes. Continued research on genetic mutations and innovative treatments offers hope for better disease management in the future.