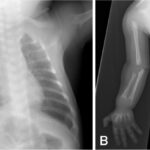
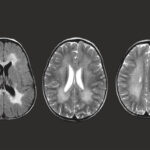
Infantile malignant osteopetrosis (IMO) is a rare genetic disorder characterized by excessively dense bones due to defective bone resorption. This condition, also known as malignant infantile osteopetrosis, severely impairs bone marrow function and can be life-threatening if untreated.
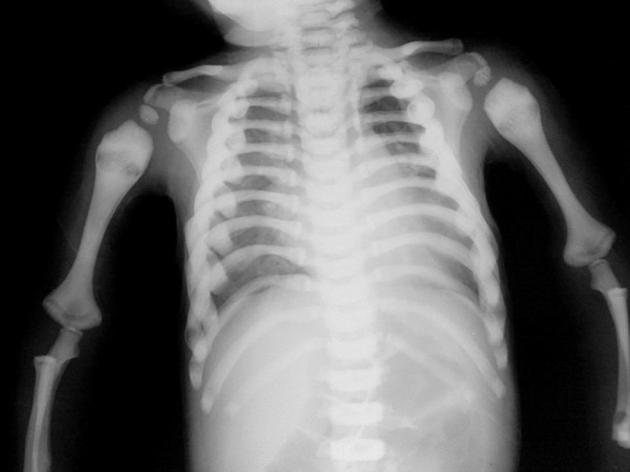
Causes of Infantile Malignant Osteopetrosis
IMO is primarily caused by mutations in genes that regulate osteoclast function, the cells responsible for bone resorption. Common gene mutations include:
- TCIRG1 (most prevalent)
- CLCN7
- OSTM1
- PLEKHM1
These mutations impair osteoclast development or activity, resulting in abnormal bone density and related complications.
Symptoms of Infantile Malignant Osteopetrosis
Symptoms typically manifest in infancy and may include:
- Failure to thrive
- Pale skin and fatigue (due to bone marrow suppression)
- Frequent infections
- Neurological issues such as hearing loss, vision impairment, and developmental delays
- Facial paralysis
- Enlarged liver and spleen (hepatosplenomegaly)
- Fractures and bone deformities
Diagnosis of Infantile Malignant Osteopetrosis
Accurate diagnosis requires:
- X-rays revealing abnormally dense bones
- Genetic testing to identify specific mutations
- Bone marrow biopsy to assess marrow function
Treatment Options for Infantile Malignant Osteopetrosis
Effective treatment strategies include:
1. Hematopoietic Stem Cell Transplantation (HSCT)
- The only curative treatment that restores normal osteoclast function.
- Early transplantation significantly improves survival rates and reduces neurological damage.
2. Supportive Therapies
- Calcium chelation therapy to reduce calcium accumulation
- Vitamin D supplements to stimulate residual osteoclast activity
- Erythropoietin therapy to manage anemia
3. Surgical Interventions
- Optic nerve decompression for vision preservation in patients with optic nerve compression.
Prognosis and Life Expectancy
Without treatment, infantile malignant osteopetrosis often leads to death in early childhood due to bone marrow failure and related complications. Successful HSCT can improve life expectancy and quality of life, although neurological damage may persist if treatment is delayed.
Prevention and Genetic Counseling
Since IMO is a genetic disorder, genetic counseling is crucial for families with a history of osteopetrosis. Carrier screening and prenatal diagnosis can help assess risks in future pregnancies.
Understanding Bone Remodeling in IMO
The imbalance between bone formation and bone resorption is the core issue in IMO. The following diagram illustrates this imbalance:
FAQs
Q1: What is the life expectancy for patients with infantile malignant osteopetrosis?
A: Without treatment, life expectancy is very limited, often within the first decade. Successful HSCT improves prognosis significantly.
Q2: Can infantile malignant osteopetrosis be detected before birth?
A: Yes, genetic testing can identify mutations in at-risk families through prenatal diagnosis.
Q3: What are the long-term effects of infantile malignant osteopetrosis?
A: Even after HSCT, patients may experience visual, hearing, or developmental impairments if the condition was untreated for an extended period.
Q4: Is there a cure for infantile malignant osteopetrosis?
A: HSCT is the only known curative treatment, but success depends on early diagnosis and intervention.
Q5: How common is infantile malignant osteopetrosis?
A: It is extremely rare, with an estimated incidence of 1 in 200,000 to 300,000 live births.
Infantile malignant osteopetrosis is a severe genetic condition that requires prompt diagnosis and treatment to improve outcomes. With advancements in HSCT and supportive therapies, the prognosis for affected children has improved significantly. Early intervention remains critical in preventing irreversible complications.