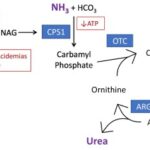
N-Acetylglutamate Synthase (NAGS) deficiency is a rare autosomal recessive metabolic disorder that disrupts the urea cycle. This condition results in the accumulation of ammonia in the blood, a condition known as hyperammonemia. Left untreated, this disorder can lead to severe neurological damage, coma, or even death.
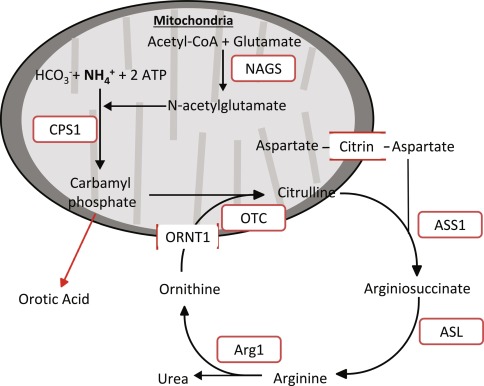
Understanding the Role of NAGS in the Urea Cycle
NAGS is a crucial enzyme responsible for synthesizing N-acetylglutamate (NAG), an essential activator of carbamoyl phosphate synthetase 1 (CPS1), the first enzyme in the urea cycle. Without adequate NAG production, CPS1 cannot efficiently convert ammonia into urea, leading to toxic ammonia buildup.
Causes of Hyperammonemia in NAGS Deficiency
NAGS deficiency is caused by mutations in the NAGS gene, which is located on chromosome 17. These mutations impair enzyme activity, reducing NAG production and compromising urea cycle functionality.
Genetic Inheritance
- Autosomal recessive pattern: Individuals must inherit two mutated copies of the NAGS gene (one from each parent) to develop the disorder.
Symptoms of Hyperammonemia in NAGS Deficiency
Symptoms can vary significantly, presenting in either neonatal onset or late-onset forms:
Neonatal Onset
- Vomiting
- Poor feeding
- Lethargy
- Hypothermia
- Seizures
- Coma (if untreated)
Late-Onset Form
- Chronic vomiting
- Developmental delay
- Behavioral disturbances
- Periodic episodes of confusion
Diagnosis of NAGS Deficiency
Accurate diagnosis involves a combination of clinical evaluation, biochemical testing, and genetic analysis:
Biochemical Tests
- Elevated plasma ammonia levels
- Normal or low plasma citrulline
- Absence of elevated orotic acid (distinguishing it from other urea cycle defects)
Genetic Testing
- Identifies mutations in the NAGS gene to confirm the diagnosis.
Treatment Options for NAGS Deficiency
Effective management aims to control ammonia levels and prevent neurological damage.
Pharmacological Treatment
- Carglumic acid (Carbaglu®): A structural analog of NAG that activates CPS1, bypassing the defective NAGS enzyme.
Dietary Management
- Protein-restricted diet: Reduces ammonia production.
- Essential amino acid supplementation: Prevents nutrient deficiencies.
Emergency Management
- Intravenous medications to lower ammonia levels rapidly
- Hemodialysis in severe hyperammonemic crises
Prognosis and Long-Term Outlook
With early diagnosis and proper management, patients with NAGS deficiency can lead relatively normal lives. Lifelong adherence to treatment protocols is crucial to minimize ammonia accumulation and prevent neurological complications.
Research and Future Directions
Ongoing research aims to improve gene therapies and novel pharmacological agents that could enhance the management of NAGS deficiency. Early newborn screening initiatives may also enhance early detection and improve outcomes.
FAQs
What triggers hyperammonemia in NAGS deficiency?
Triggers include excessive protein intake, infections, prolonged fasting, and stress, all of which increase ammonia production.
Can NAGS deficiency be cured?
Currently, there is no cure, but treatments like Carglumic acid can effectively manage ammonia levels and improve quality of life.
How common is NAGS deficiency?
NAGS deficiency is extremely rare, with only a few hundred reported cases worldwide.
Is NAGS deficiency fatal?
If untreated, severe hyperammonemia can be life-threatening. However, early intervention significantly improves prognosis.
How is neonatal NAGS deficiency diagnosed?
Newborns presenting with severe hyperammonemia undergo biochemical tests and genetic screening to confirm NAGS deficiency.