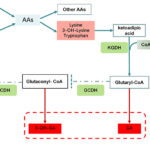
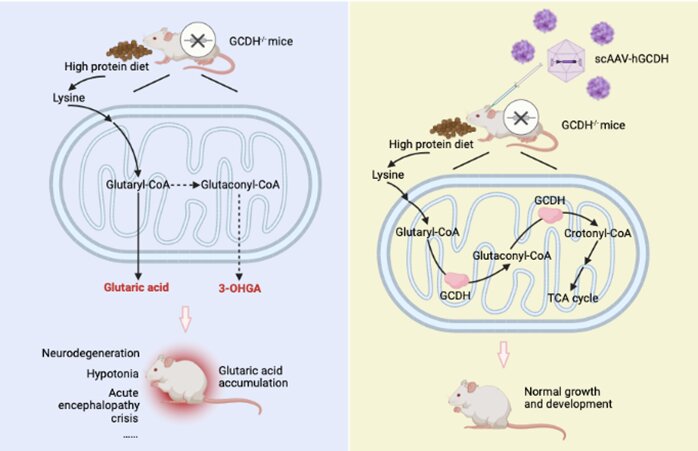
Glutaric Aciduria Type II (GA-II), also known as Multiple Acyl-CoA Dehydrogenase Deficiency (MADD), is a rare inherited metabolic disorder characterized by impaired fatty acid, amino acid, and choline metabolism. This condition is part of a broader group of mitochondrial disorders affecting energy production.
Causes of Glutaric Aciduria Type II
GA-II is caused by mutations in genes responsible for electron transfer proteins essential for fatty acid oxidation. The following genes are commonly implicated:
- ETFA (Electron Transfer Flavoprotein Alpha Subunit)
- ETFB (Electron Transfer Flavoprotein Beta Subunit)
- ETFDH (Electron Transfer Flavoprotein Dehydrogenase)
These mutations impair mitochondrial function, leading to toxic accumulation of organic acids and resulting in severe metabolic complications.
Symptoms of Glutaric Aciduria Type II
GA-II presents with varied symptoms depending on the onset and severity. The three primary clinical presentations include:
Neonatal Onset (Severe Form)
- Hypotonia (muscle weakness)
- Respiratory distress
- Hepatomegaly (enlarged liver)
- Hypoglycemia (low blood sugar)
- Cardiomyopathy
- Distinctive odor (similar to sweaty feet)
Infantile Onset
- Developmental delays
- Vomiting and feeding difficulties
- Metabolic crises triggered by illness or fasting
- Muscle weakness
Late-Onset (Milder Form)
- Muscle pain
- Exercise intolerance
- Progressive myopathy
Diagnosis of Glutaric Aciduria Type II
Diagnostic approaches involve a combination of clinical evaluation, biochemical testing, and genetic analysis. Key diagnostic methods include:
- Blood Tests: Elevated acylcarnitines and organic acids
- Urine Analysis: Presence of glutaric acid, ethylmalonic acid, and other organic acids
- Genetic Testing: Identifies mutations in ETFA, ETFB, or ETFDH genes
Treatment Options for Glutaric Aciduria Type II
While there is no cure for GA-II, management strategies aim to control symptoms and prevent metabolic crises. Key treatment approaches include:
Dietary Management
- High-Carbohydrate, Low-Fat Diet to minimize fatty acid metabolism
- Avoidance of Prolonged Fasting to reduce metabolic stress
- Supplementation with riboflavin (vitamin B2), carnitine, and coenzyme Q10
Medical Interventions
- IV Glucose Infusions during metabolic crises
- Emergency Protocols for acute metabolic episodes
Long-Term Management
- Regular monitoring of metabolic markers
- Early intervention to prevent complications
Prognosis of Glutaric Aciduria Type II
Prognosis varies based on disease severity. Neonatal-onset forms often have a poor outlook, whereas individuals with late-onset GA-II can achieve improved quality of life with appropriate management.
Preventive Measures
- Genetic Counseling for at-risk families
- Newborn Screening programs can facilitate early detection and treatment
FAQs
What triggers metabolic crises in Glutaric Aciduria Type II?
Metabolic crises are often triggered by fasting, illness, or increased energy demand. Maintaining a regular feeding schedule and managing infections promptly can help reduce the risk.
Is Glutaric Aciduria Type II hereditary?
Yes, GA-II follows an autosomal recessive inheritance pattern. Both parents must carry a defective gene for the condition to manifest in their child.
Can Glutaric Aciduria Type II be detected at birth?
Yes, newborn screening programs can identify markers for GA-II, enabling early intervention and improved outcomes.
What is the role of riboflavin in managing GA-II?
Riboflavin acts as a cofactor in the electron transport chain, improving energy production and metabolic stability in some patients.
Are there any lifestyle adjustments required for GA-II patients?
Patients are advised to avoid fasting, maintain a high-carbohydrate diet, and follow a structured meal plan to prevent metabolic imbalances.
Glutaric Aciduria Type II is a complex metabolic disorder requiring early diagnosis and comprehensive management strategies. With prompt intervention, individuals with milder forms of the condition can achieve better long-term outcomes.