Glutaric aciduria type I (GA-I) is a rare genetic metabolic disorder characterized by the body’s inability to process certain amino acids properly. This condition leads to the accumulation of glutaric acid and related compounds, which can cause severe neurological damage if untreated. GA-I is typically identified in infancy or early childhood and requires early intervention to prevent serious complications.
Causes of Glutaric Aciduria Type I
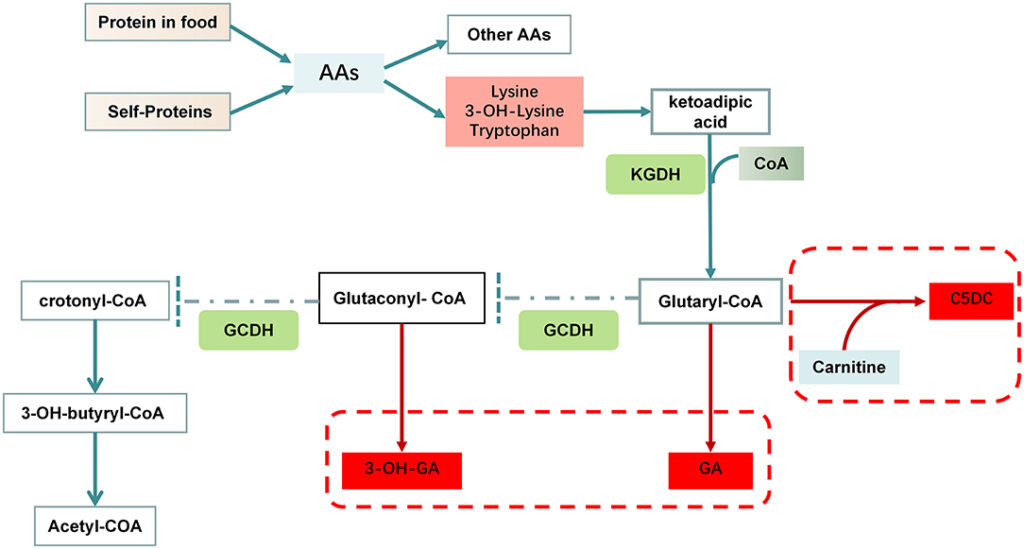
GA-I is caused by mutations in the GCDH gene, which encodes the enzyme glutaryl-CoA dehydrogenase. This enzyme is essential for breaking down the amino acids lysine, hydroxylysine, and tryptophan. Mutations in the GCDH gene impair this metabolic pathway, leading to the accumulation of toxic byproducts such as glutaric acid, 3-hydroxyglutaric acid, and glutaryl-CoA in bodily tissues.
Inheritance Pattern
GA-I follows an autosomal recessive inheritance pattern. This means that an affected individual must inherit two defective copies of the GCDH gene (one from each parent) to develop the disorder. Carriers, who possess only one mutated gene, typically show no symptoms.
Symptoms of Glutaric Aciduria Type I
The symptoms of GA-I vary, but common signs include:
- Macrocephaly (abnormally large head)
- Hypotonia (low muscle tone)
- Developmental delays
- Dystonia (involuntary muscle contractions)
- Seizures
- Progressive brain atrophy
During metabolic crises, triggered by illness or fasting, individuals may experience acute neurological deterioration, which can result in irreversible damage.
Diagnosis of Glutaric Aciduria Type I
Early diagnosis is crucial to prevent severe complications. Diagnostic procedures include:
- Newborn Screening: Standard in many countries, this test detects elevated glutaric acid levels.
- Urine Organic Acid Analysis: Identifies elevated glutaric acid concentrations.
- Plasma Amino Acid Testing: Measures abnormal amino acid levels.
- Genetic Testing: Confirms mutations in the GCDH gene.
- Brain Imaging (MRI/CT): Often reveals characteristic basal ganglia atrophy or widened cerebrospinal fluid spaces.
Treatment and Management
Treatment strategies aim to reduce glutaric acid buildup and prevent metabolic crises. Key approaches include:
1. Dietary Management
- Low-protein diets that restrict lysine and tryptophan intake
- Special metabolic formulas providing essential nutrients without harmful amino acids
- Supplementation with L-carnitine to enhance energy production and reduce toxic metabolites
2. Medication
- Riboflavin (Vitamin B2) may enhance residual enzyme activity in some cases
- Emergency medications to manage acute metabolic crises
3. Routine Monitoring
- Regular metabolic assessments and neurological evaluations are essential for early intervention and to track disease progression.
Prognosis and Long-Term Outlook
With early diagnosis and proper management, many children with GA-I can achieve normal development and avoid neurological damage. However, untreated cases often result in severe disability or early mortality.
Preventive Strategies
While genetic conditions like GA-I cannot be prevented outright, proactive steps can minimize risks:
- Prenatal Genetic Counseling: Advisable for families with a known history of GA-I
- Carrier Screening: Identifies at-risk individuals before conception
- Newborn Screening Programs: Enable early detection and intervention
Glutaric aciduria type I is a manageable condition when diagnosed early and treated properly. Through dietary regulation, medication, and continuous medical monitoring, affected individuals can achieve improved outcomes and avoid severe neurological complications.