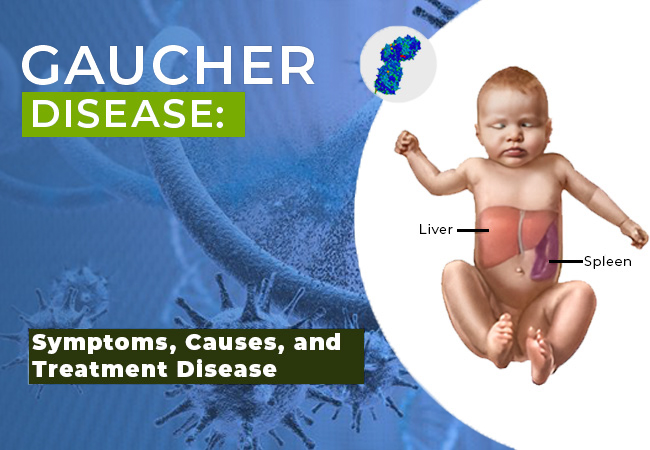
Gaucher’s disease is a rare genetic disorder caused by a deficiency of the enzyme glucocerebrosidase. This leads to the accumulation of glucocerebroside in the spleen, liver, and bone marrow, resulting in organ enlargement, bone abnormalities, and other systemic complications. It is classified as a lysosomal storage disorder and follows an autosomal recessive inheritance pattern.
Types of Gaucher’s Disease
Gaucher’s disease is divided into three main types based on severity and neurological involvement:
Type 1 (Non-Neuronopathic Gaucher’s Disease)
- Most common form, accounting for 90% of cases.
- No central nervous system involvement.
- Symptoms include hepatosplenomegaly (enlarged liver and spleen), anemia, thrombocytopenia, and bone pain.
Type 2 (Acute Neuronopathic Gaucher’s Disease)
- A rare and severe form that affects infants.
- Rapid neurological deterioration leading to death by age two.
- Symptoms include muscle rigidity, seizures, and respiratory distress.
Type 3 (Chronic Neuronopathic Gaucher’s Disease)
- Intermediate severity, with neurological and systemic symptoms.
- Affects childhood or adolescence with progressive neurological decline.
- Symptoms include cognitive impairment, ataxia, and seizures.
Causes and Genetics of Gaucher’s Disease
Gaucher’s disease is caused by mutations in the GBA gene, responsible for producing the enzyme glucocerebrosidase. This enzyme is essential for breaking down glucocerebroside, a fatty substance. The disorder follows an autosomal recessive inheritance, meaning both parents must carry a mutated gene for their child to inherit the condition.
Symptoms of Gaucher’s Disease
Symptoms vary based on the type and severity of the disease but commonly include:
- Hepatosplenomegaly: Enlarged liver and spleen causing abdominal distension.
- Anemia and Thrombocytopenia: Low red blood cells and platelets leading to fatigue and easy bruising.
- Bone Pain and Fragility: Osteoporosis and avascular necrosis (bone death) leading to fractures.
- Neurological Issues (Type 2 & 3): Developmental delays, seizures, and motor dysfunction.
Diagnosis of Gaucher’s Disease
Accurate diagnosis involves multiple laboratory and imaging tests:
Blood Tests and Enzyme Assay
- Measures glucocerebrosidase enzyme activity in white blood cells.
- Low enzyme levels confirm the disease.
Genetic Testing
- Identifies mutations in the GBA gene.
- Confirms carrier status in family members.
Imaging Studies
- MRI and CT Scans: Assess liver and spleen size.
- X-rays and Bone Scans: Detect bone damage and osteoporosis.
Treatment Options for Gaucher’s Disease
While there is no cure, various treatments help manage symptoms and improve quality of life.
1. Enzyme Replacement Therapy (ERT)
- Standard treatment for Type 1 and mild Type 3 Gaucher’s disease.
- Administers recombinant glucocerebrosidase intravenously.
- Drugs include Imiglucerase, Velaglucerase alfa, and Taliglucerase alfa.
2. Substrate Reduction Therapy (SRT)
- Reduces glucocerebroside accumulation by inhibiting its production.
- Includes Eliglustat and Miglustat, used for Type 1 Gaucher’s disease.
3. Bone Marrow Transplant
- Rarely performed but can provide a long-term cure.
- Replaces diseased marrow with healthy donor marrow.
4. Supportive Care
- Pain Management: Use of analgesics and anti-inflammatory medications.
- Blood Transfusions: For anemia and low platelet count.
- Physical Therapy: To maintain mobility and reduce bone pain.
Living with Gaucher’s Disease
Patients can manage the condition effectively with early diagnosis and consistent treatment. Lifestyle modifications include:
- Regular Monitoring: Routine check-ups with a hematologist and genetic specialist.
- Diet and Nutrition: A balanced diet rich in calcium and vitamin D for bone health.
- Physical Activity: Low-impact exercises such as swimming or yoga.