Deficiency of Interleukin-1 Receptor Antagonist (DIRA) is an uncommon autoinflammatory disease resulting from autosomal recessive mutations in the IL1RN gene. This condition leads to unregulated activity of interleukin-1 (IL-1), causing systemic inflammation with prominent skin and bone manifestations. Early recognition and appropriate management are crucial to prevent severe complications and mortality.

Genetic Basis and Pathophysiology
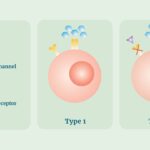
DIRA is caused by loss-of-function mutations in the IL1RN gene, which encodes the interleukin-1 receptor antagonist (IL-1Ra). IL-1Ra is a natural inhibitor of IL-1, a cytokine involved in inflammatory responses. In the absence of functional IL-1Ra, IL-1 signaling remains unchecked, leading to excessive inflammation. This unopposed IL-1 activity manifests as severe systemic inflammation, particularly affecting the skin and bones.
Clinical Manifestations
Patients with DIRA typically present in the neonatal period or early infancy with the following features:
- Skin Involvement: Generalized pustulosis characterized by widespread sterile pustules, erythema, and scaling. The skin lesions can exacerbate with minor trauma or interventions, such as intravenous cannula insertion.
- Bone Involvement: Multifocal osteomyelitis presenting as painful bony swellings, often with overlying erythema and warmth. Radiological findings may reveal periosteal elevation and mixed lytic-sclerotic lesions. Notably, the inflammation primarily involves the periosteum, sparing the joint spaces.
- Systemic Inflammation: Elevated acute phase reactants, including increased erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). Patients may also exhibit irritability, poor feeding, and failure to thrive due to chronic pain and systemic illness.
Diagnosis
The diagnosis of DIRA involves a combination of clinical evaluation, laboratory investigations, and genetic testing:
- Clinical Assessment: Recognition of the characteristic skin and bone manifestations, along with a history of early-onset systemic inflammation.
- Laboratory Findings: Elevated inflammatory markers such as ESR and CRP. Cultures from skin and bone lesions are typically sterile, ruling out infectious causes.
- Genetic Testing: Confirmation through identification of biallelic mutations in the IL1RN gene. Techniques such as single nucleotide polymorphism (SNP) array and Sanger sequencing are employed to detect deletions or point mutations.
Treatment
The cornerstone of DIRA management is the administration of recombinant IL-1Ra to counteract the unopposed IL-1 activity:
- Anakinra: A recombinant form of IL-1Ra administered via daily subcutaneous injections. Anakinra has been shown to induce rapid and sustained clinical remission, resolving skin and bone inflammation and normalizing inflammatory markers. However, some patients may develop hypersensitivity reactions, necessitating desensitization protocols.
- Rilonacept: An IL-1 inhibitor that has been approved for maintenance of remission in DIRA patients. It is administered as a weekly subcutaneous injection and may offer an alternative for patients intolerant to anakinra.
Prognosis
With early diagnosis and appropriate IL-1 blockade therapy, patients with DIRA can achieve significant clinical improvement and lead a near-normal life. Delayed diagnosis or untreated disease can result in severe complications, including growth disturbances, permanent bone deformities, and increased mortality risk in early childhood.
DIRA is a life-threatening autoinflammatory disorder arising from mutations in the IL1RN gene, leading to uncontrolled IL-1-mediated inflammation. Prompt recognition and treatment with IL-1 antagonists are essential to prevent serious outcomes. Ongoing research and increased awareness are vital to improve diagnosis and management strategies for this rare condition.